
The interest in cancer as an epigenetic, as well as a genetic disease, has markedly accelerated over the past decade. This chapter concentrates on one particularly interesting aspect of this – the growing evidence that one of the most studied epigenetic abnormalities in cancer, abnormal gene silencing associated with gene promoter DNA hypermethylation, is linked to key aspects of chromatin regulation of gene expression which maintains the state of embryonic stem (ES)/progenitor cells. This is a timely juxtaposition since there is also a growing body of data suggesting that cancer “stem/initiating cells”, especially when they may dominate in the most aggressive forms of human tumors, have a gene expression signature reminiscent of ES cells. A key part of this signature is an increase in polycomb protein group (PcG) complex constituents, and a decrease in expression of a group of genes for which PcG targeting is normally key to maintaining a poised, low basal activity, to maintain ES/progenitor cell status. A working hypothesis is explored that this primitive cell status in cancers may be, in part, maintained by a similar chromatin regulation of key genes with the addition, to many, of a fundamental abnormality, DNA methylation in CpG islands of the gene promoters. This DNA change is generally not a feature of the PcG targeted genes in ES/progenitor cells and may eliminate a pre-requisite plasticity for these genes to ensure that their activation would normally allow lineage commitment and maturation. The early appearance of the DNA hypermethylation of at least a large group of genes in cancer development could reflect a key role for locking in a primitive state of abnormally expanding cells – which then provides an important substrate for the later changes that drive progression to invasive cancer.
1. Introduction
Nowhere, in terms of disease, will the emerging knowledge of epigenetic control of gene expression patterns in embryogenesis and stem cells be more important than for our understanding of human cancer. Over the past decade, there has been a surge of interest in the concept that cancer, as well as being a disease driven by genetic alterations, is also a disease of epigenetic abnormalities (Herman and Baylin, 2003; Jones and Baylin, 2007; Feinberg and Tycko, 2004; Esteller, 2008). Overall changes in levels of key histone modifications important to activation and repression of gene transcription have been reported in tumors and the blood of patients with cancer (Fraga et al., 2005; Seligson et al., 2005). Likewise, both decreases and increases in DNA methylation have been recognized in cancer DNA over the past 20 years (Herman and Baylin, 2003; Jones and Baylin, 2007; Feinberg and Tycko, 2004; Esteller, 2008). The critical issues for research today is to derive understanding of the molecular and cell biology mechanisms contributing to these overall epigenetic changes and to crisply outline the ramifications of the changes for our basic understanding of cancer biology and the translational implications for improved care of patients with these diseases.
Of all the chromatin changes now recognized in cancer, as has been the subject of multiple recent reviews (Herman and Baylin, 2003; Jones and Baylin, 2007; Feinberg and Tycko, 2004; Esteller, 2008), none has received more scrutiny than the phenomenon of aberrant gene silencing associated with abnormal appearance of DNA methylation in the proximal promoter regions of associated genes. This methylation involves those genes with promoter region CpG islands which are largely protected from DNA methylation in all normal cells including those in cells from tissues in embryonic development and in adult cell renewing systems (Herman and Baylin, 2003; Jones and Baylin, 2007; Feinberg and Tycko, 2004; Esteller, 2008; Baylin and Ohm, 2006). Some of the genes involved are those recognized as being key tumor suppressor genes (Herman and Baylin, 2003; Jones and Baylin, 2007; Feinberg and Tycko, 2004; Esteller, 2008; Baylin and Ohm, 2006). The promoter DNA hypermethylation and gene silencing then constitute an alternative to mutations, in genes with otherwise wild type genetic constitution, for providing a loss of function which facilitates tumor initiation and progression (Herman and Baylin, 2003; Jones and Baylin, 2007; Feinberg and Tycko, 2004; Esteller, 2008; Baylin and Ohm, 2006).
In addition to involving such above classic tumor suppressor genes, it is increasingly recognized that many genes with an infrequent, or no, history of mutations may frequently be altered in cancer through the above gene silencing mechanism (Jones and Baylin, 2007). Thus, many genes which would otherwise not be recognized as important for loss of function in cancer development are being discovered by searching for their involvement with abnormal promoter DNA methylation in tumor DNA (Yamashita et al., 2002; Schuebel et al., 2007; Smiraglia et al., 2007). In fact, there is a growing enterprise for randomly searching cancer genomes for such changes as now represented by work ongoing in the Cancer Genome Atlas project or (TCGA, 2008). Such searches are enriching our knowledge of how cancers develop and progress and are providing new tools with high potential for improving cancer diagnosis and management (TCGA, 2008).
In the present chapter, the above changes in promoter DNA methylation are reviewed within the special context of their recently recognized relationships to chromatin patterns which are essential for the regulation of gene expression in normal embryonic stem (ES) and progenitor cells. The implications for these relationships, and of the genes involved, for how human cancers evolve will be a focus. A working construct will be emphasized, extending previous discussions (Baylin and Ohm, 2006; Ohm and Baylin, 2007), which proposes that cancers often evolve within the context of stress responses to chronic injury which foster the abnormal expansion of cells with ES and progenitor cell features. For these cells, epigenetic abnormalities are essential for their survival and growth and for their responsiveness to subsequent genetic and epigenetic changes which drive tumor progression.
2. Relationships bewteen promoter DNA hypermethylation, genes which regulate stem/progenitor cells, and initiation of human cancer
The leading risk factors for the most prevalent forms of human cancer involve settings such as aging and chronic inflammation and which induce chronic increases in attempted cell renewal (Balkwill and Coussens, 2004; Nelson et al., 2004; Lu et al., 2006). We have suggested how epigenetic changes in cells, in these settings, might be essential to protect cells from toxicities resulting from build up in DNA damaging agents such as reactive oxygen species (ROS) and stimuli which can normally challenge cells to enter apoptosis, or senescence (Baylin and Ohm, 2006; Ohm and Baylin, 2007). Thus, cancer risk states are actually antithetical to the abnormal cell expansion which arises in these scenarios and which then, over periods of years, foster neoplasia. By definition, cells must evolve survival mechanisms which allow their participation in abnormal cell expansion and evolution of pre-invasive lesions. Certainly, key gene mutations can play a role in this survival by addicting cells to overactivity of key developmental and signal transduction pathways, such as Wnt (Baylin and Ohm, 2006; Taipale and Beachy, 2001; Gregorieff and Clevers, 2005; Ben-Porath et al., 2008; Hanahan and Weinberg, 2000) sonic hedgehog (Taipale and Beachy, 2001; Berman et al., 2003), and myc signaling (Ben-Porath et al., 2008; Hanahan and Weinberg, 2000), which function for maintenance of normal stem/progenitor cell states in development, and which often drive tumorigenesis. However, simple introduction of such mutations into mature cells often triggers apoptosis or senescence (Collado et al., 2007; Collado et al., 2005; Lowe et al., 2004) suggesting that other cell changes may be essential for the mutations to play their tumorigenic role.
We suggest that gene promoter DNA hypermethylation and abnormal gene silencing may be one key cellular change which can allow cells to survive the toxic surroundings of cancer risk states and take advantage of key gene mutations (Jones and Baylin, 2007; Baylin and Ohm, 2006; Ohm and Baylin, 2007). These changes can allow cells to resist death normally triggered by DNA damage and to utilize key gene mutations as a selective advantage for survival and growth rather than signals for apoptosis and/or senescence (Jones and Baylin, 2007; Baylin and Ohm, 2006; Ohm and Baylin, 2007).
What is the evidence for the above hypothesis? At least two features of the abnormal gene DNA methylation during tumor progression are strongly consistent with such a proposal. First, this promoter change can involve many key genes involved in maintenance of stem/progenitor populations in embryonic development and adult cell renewal (Jones and Baylin, 2007; Baylin and Ohm, 2006). Second, the abnormal methylation frequently arises, for such genes, early in neoplastic evolution before the appearance of frank invasive phases of common forms of human cancer (Jones and Baylin, 2007; Baylin and Ohm, 2006). Thus, as illustrated for the progression of colon cancer in Figure 1, epigenetically mediated loss of function for key genes, such as p16 for cell cycle control, transcription factors such as the GATA4- and -5 factors, and repressive genes for the Wnt pathway (SFRP’s and SOX17), often occurs concordantly in pre-malignant lesions.
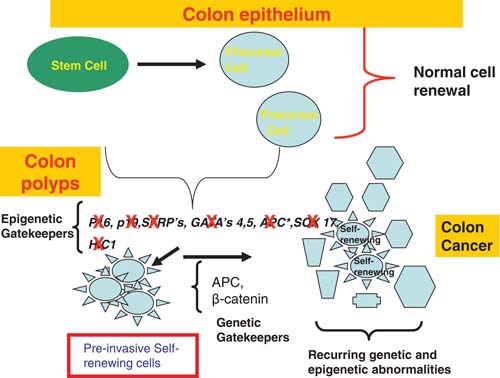
The top panel shows that normal stem cells in adult intestinal cell epithelial renewal utilize the genes listed as epigenetic gatekeepers to control stem/progenitor cell proliferation and generate precursor cells which mature normally. When these genes are abnormally silenced, especially in groups, as shown in the colon polyps in the middle panel, this helps foster, abnormal, pre-invasive stem/progenitor cell expansion (red box showing neoplastic self-renewing cells), creating polyps and a cancer risk state. The risk state, as per the text, can foster oncogenic responses to key mutations which are labeled as the genetic gatekeepers. This combination of epigenetic and genetic abnormalities, and other recurring ones, can lead to invasive colon cancers built upon the self-renewing cells originally present and new ones created by the combination of molecular changes.
These epigenetic abnormalities for pathways important to stem/progenitor cell control in early stages of neoplastic development are extremely well illustrated for overactivity of the Wnt pathway in colon tumorigenesis (see Figure 2). Multiple genes, which can normally inhibit activation of this pathway, undergo, often in concert, early abnormal DNA methylation in pre-invasive, risk stages, of colon cancer (Suzuki et al., 2004; Zhang et al., 2008). The proteins encoded, which would be diminished or lost with the abnormal gene silencing (Finch et al., 1997), normally work all the way from the cell membrane to the nucleus to interfere with Wnt activation of β-catenin, the key mediator of canonical Wnt pathway transcriptional readout (Reya and Clevers, 2005). Thus, at the cell membrane, the family of secreted frizzled genes (SFRP’s), WIF-1, and DKK-1, multiple of which are virtually always DNA hypermethylated in colon pre-cancerous lesions (Finch et al., 1997), block Wnt interaction with its surface receptors (Finch et al., 1997; Rattner et al., 1997). This results in activation of the cytoplasmic APC complex (Finch et al., 1997) which then promotes phosphorylation of cytoplasmic β-catenin, thus targeting it for proteosomal degradation (Reya and Clevers, 2005).
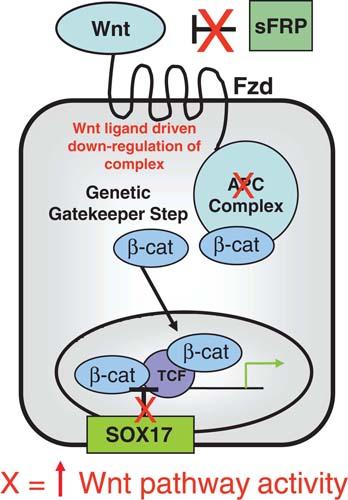
Three levels of early epigenetic events for gene silencing (red X = resulting in contributions to Wnt pathway activation) are shown. SFRP family genes, which normally help prevent Wnt proteins from interacting the their Frizzled receptors, are an example of cell membrane Wnt receptors that are frequently abnormally silenced and help lead to constitutive Wnt signaling, resulting in down regulation of the APC complex, starting at the cell membrane. This begins to decrease β-catenin degradation, resulting in increased cytoplasmic levels of this protein. While most tumors, at least among colon cancers, inactivate the APC complex through mutations in the APC gene, or through activating mutations in β-catenin, occasional tumors epigentically inactivate APC. Finally, silencing of the SOX17 gene, as per the text, results in loss of a block to TCF- β-catenin interaction helping to lead to use of the increased cellular β-catenin to activate the Wnt pathway activity. All of this concordant abnormal gene silencing primes cells for oncogenic response to key mutations, such as the APC mutations or activating mutations in β-catenin.
SOX17 is our most recently defined gene for regulation of Wnt pathway activity found to be virtually, universally, DNA hypermethylated in benign colon polyps (Zhang et al., 2008). Loss of function for this gene is a vivid example for an epigenetically mediated abnormality in stem/progenitor cell related control. Sox17 is an important member of the Sox family, which is ubiquitous in the animal kingdom and involved in diverse developmental processes including germ layer formation, cell-type specification, and organogenesis (Wegner, 1999). The importance of Sox17 for embryonic colon development has been demonstrated by two knock-out experiments wherein Sox17-null mouse embryos exhibit a deficiency of maturation of gut definitive endoderm, which leads to embryonic lethality before day E10.5 (Kanai-Azuma et al., 2002). Unlike for the cell membrane associated, anti-Wnt factors discussed earlier, SOX17 appears to block the Wnt pathway in the nucleus (Sinner et al., 2007; Sinner et al., 2004; Zorn et al., 1999). Recent studies suggests that xenopus and mouse Sox17 suppress canonical Wnt signaling by competing with the interaction of β-catenin with its transcriptional activation partners, TCF/LEF (Sinner et al., 2007; Sinner et al., 2004; Zorn et al., 1999). Another group (Sinner et al., 2007), and our recent studies (Zhang et al., 2008), have validated that SOX17 inhibits, in human colon cancer cells, endogenous TCF/β-catenin-mediated transcription. This activity requires, in our studies, a well conserved N-terminal HMG box domain (Zhang et al., 2008).
3. Mouse knockout studies of the gene, hypermethylated in cancer 1 (HIC1), indicate that stem/progenitor cell related, epigenetic abnormalities, can contribute to the initiation of cancer
There are recent studies in mouse models indicating that over-expression of ES cell transcription factors (Ben-Porath et al., 2008) and loss of proper DNA methylation associated control of gene imprinting can lead to stem/progenitor cell expansion and contribute to the initial stages of cancer (Baylin and Ohm, 2006; Ohm and Baylin, 2007; Feinberg et al., 2006; Sakatani et al., 2005; Ohm et al., 2007). With specific respect to the DNA hypermethylated cancer genes under discussion in this chapter, in our work, we have developed a mouse model for disruption of the gene Hic1. We first identified human Hic1 in a random screen for DNA hypermethylated cancer genes (Wales et al., 1995) residing on chromosome 17p13, a frequently targeted locus for allelic loss in human tumors (Wales et al., 1995). This gene very frequently undergoes CpG island hypermethylation and silencing in pre-invasive and invasive forms of virtually all common forms of human cancer (Wales et al., 1995; Chen et al., 2005; Chen et al., 2003; Chen et al., 2004). The role for Hic1 in development became apparent when our constitutive knockout of this gene proved to be embryonic lethal (Carter et al., 2000). Homozygous embryos show a variable phenotype, ranging from severe craniofacial, neural and other defects (Carter et al., 2000).
Despite the lethality of the homozygous knock state of Hic1-/- mice, heterozygous animals (Hic1± mice) have an age dependent predisposition to epithelial cancers in males and to lymphomas and soft issue sarcomas in females (Chen et al., 2003; Chen et al., 2004). Crosses of Hic1± mice with p53 ± mice showed cooperative loss of the two genes, yielding distinct tumor phenotypes involving breast and ovarian carcinomas and metastatic osteosarcomas (Chen et al., 2004). Importantly, in all of the above mouse settings, there is always retention of the wild type copy of Hic1 in tumors with loss of its function through promoter hypermethylation and associated gene silencing (Chen et al., 2003; Chen et al., 2004). Mouse knockout of Hic1 thus provides, in effect, a model for one abnormal, epigenetic step to tumorigenesis. For this abnormality, we have previously reviewed how loss of HIC1 function can potentially set off a network of tumorigenic effects, expounded upon below, including augmentation of other epigenetic gene silencing events (Jones and Baylin, 2007; Chen et al., 2005).
Hic1 appears linked to regulation of embryonic development and tumorigenesis via its master role as a transcription factor and its membership in the POZ domain family of Zinc finger containing transcriptional repressors (Bardwell and Treisman, 1994). Our lab and collaborators have now, to date, identified two such direct repressor targets for Hic1, both important for embryogenesis, atonal and SIRT1 (Chen et al., 2005; Briggs et al., 2008). SIRT1 is a key cell survival protein in lower organisms and mammalian cells (Kaeberlein et al., 1999; Lin et al., 2000; Cohen et al., 2004; Lim, 2006) which is increased several fold in cells from Hic1-/- mice and tumors evolving in Hic1± mice (Bardwell and Treisman, 1994). As a protein which functions in gene silencing from yeast to mammals (Motta et al., 2004; Pruitt et al., 2006; Kimura et al., 2002; Suka et al., 2002), we have tied promoter localization of SIRT1 to the silencing of cancer genes affected by promoter DNA hypermethylation (Pruitt et al., 2006). Finally, increased levels of SIRT1 in the setting of silencing of HIC1 can interfere with the transcriptional function of the key tumor suppressor, p53 (Chen et al., 2005).
Most recently, the role of epigenetic loss of Hic1 in early tumorigenes, has been linked by Watkins and colleagues in a mouse model to its transcriptional repression of the gene, Atonal (ATOH1) (Briggs et al., 2008). In this study, Hic1 was identified as a direct transcriptional repressor of Atonal Homolog 1 (Atoh1), a proneural transcription factor essential for function of granule cell precursors (GCPs) and cerebellar development (Ben-Arie et al., 1997; Ben-Arie et al., 2000). Medulloblastoma is an embryonal tumor thought to arise from such cells and to be driven, in part by sonic hedgehog signaling (Kenney et al., 2003; Berman et al., 2002; Kenney and Rowitch, 2000). PATCHED (PTCH) is an inhibitor of Hedgehog signaling and is one tumor suppressor gene important for medulloblastoma development, being mutated in 20% or less of these tumors (Briggs et al., 2008; Berman et al., 2002). In addition, 17p13.3 locus deletions, the site of HIC1, are the most common genetic alteration in medulloblastoma – and the gene is frequently DNA hypermethylated and silenced in this tumor type (Wales et al., 1995; Rood et al., 2002). When crossed with Ptch1 heterozygous mutant mice, Ptch1/Hic1 heterozygote mice display a fourfold increase in medulloblastoma and ATOH1 expression was demonstrated to be required for human medulloblastoma cells (Briggs et al., 2008). As in the studies previously detailed for tumors in Hic1± mice, the wild type copy of the gene was retained, and its promoter DNA hyper-methylated, in medulloblastomas from the Hic1±,Ptch± mice (Briggs et al., 2008). These studies squarely model the potential for abnormal epigenetic gene silencing to participate in expansion of progenitor cells and early steps in tumorigenesis.
4. Relationships between a regulatory chromatin pattern in embryonic cells, abnormal promoter DNA hypermethylation, and cell progression to cancer
Recent studies by our group (Ohm and Baylin, 2007; Ohm et al., 2007; McGarvey et al., 2008), and others (Widschwendter et al., 2007; Schlesinger et al., 2007), of the chromatin involved with the promoters of genes with DNA hypermethylation in cancer, have provided yet more strong links between embryonic cells and epigenetic abnormalities arising in human neoplasia. The key molecular players in the relationships are the polycomb group of proteins (PcG) which interact to maintain long term gene transcriptional repression in development from model organisms to mammals and the trithorax (Trx) proteins which similarly maintain active gene transcription (Boyer et al., 2006; Orlando, 2003; Ringrose, 2006; Lund AavL, 2004; Valk-Lingbeek et al., 2004). It is first important to discuss key elements of regulation for genes which are essential for maintenance of ES cells and embryonic progenitor cells and for which expression is controlled by a balance in activity of PcG and Trx at their promoters. This balance defines a promoter chromatin state termed “bivalent chromatin” (Bernstein et al., 2006; Chi and Bernstein, 2009; Mikkelsen et al., 2007). This is defined as the simultaneous presence of a narrow zone, around the start sites of such genes, of the Trx mediated histone modification, methylation of lysine 4 of histone H3 (H3K4me), and a broader surrounding zone of a repressive mark, H3K27me3, established by the PcG proteins (Bernstein et al., 2006; Chi and Bernstein, 2009; Mikkelsen et al., 2007). This balance is associated primarily with promoter CpG island containing genes and, importantly, serves to establish a low, poised transcription state of involved genes (Bernstein et al., 2006; Chi and Bernstein, 2009; Mikkelsen et al., 2007). This state of low expression, in turn, appears essential for prevention of premature lineage commitment of ES or embryonic progenitor cells during development (Bernstein et al., 2006; Chi and Bernstein, 2009; Mikkelsen et al., 2007). The bivalent chromatin can be remodeled to an active state, with dominance of the H3K4me mark for genes for which expression is required in more committed cells of given lineages (Bernstein et al., 2006; Chi and Bernstein, 2009; Mikkelsen et al., 2007).
Importantly, none of the normal regulatory events discussed just above, in either embryonic or committed cell settings, involve the presence of promoter DNA methylation (Mikkelsen et al., 2008). How, then, do the above concepts relate to the cancer specific DNA hypermethylation of genes? First, our group (Ohm et al., 2007; McGarvey et al., 2008), and two others (Widschwendter et al., 2007; Schlesinger et al., 2007), recognized that an inordinate percentage (50%) of genes which are DNA hypermethylated in colon cancer were found to be among the ∼10% of genes, found in tiling array studies of others, to have PcG constituents at their promoters in either ES cells or embryonic mesenchymal and neural progenitor cells. Many of these genes are also among those marked by bivalent chromatin, and low, poised expression, in these embryonic cells. Further, links between the above embryonic chromatin patterns and genes which are DNA hypermethylated in cancer have emerged from local chromatin immunoprecipation (ChIP) studies of such genes in breast and colon cancer, and subsequent ChIP-chip tiling arrays of colon cancer cells (McGarvey et al., 2008; McGarvey et al., 2006). Each of the genes examined, revealed that in the DNA hypermethylated state, the gene promoters had a series of repressive chromatin marks which included the PcG mark, H3K27me3 (McGarvey et al., 2008; McGarvey et al., 2006). Most important, when these genes were induced to DNA demethylate, and exhibit some re-expression, by either cell treatment with the DNA demethylating agent, 5-deoxy-azacytidine (DAC) (McGarvey et al., 2006), or in colon cancer cells with genetic knockout of DNA methyltransferases (McGarvey et al., 2008; McGarvey et al., 2006), the repressive marks including H3K27me3 remained and even increased, even as the active mark, H3K4me2, also increased. This state (see Figure 3) resembles bivalent chromatin.
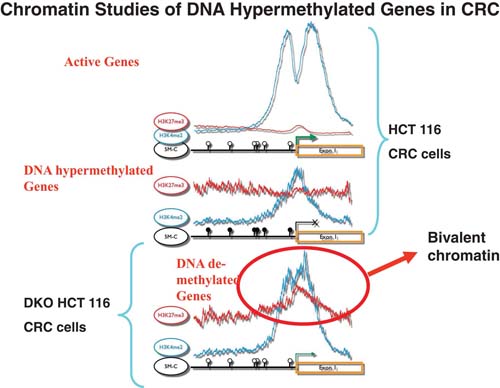
The results from a previous publication (McGarvey et al., 2008) are summarized here to reflect what has been learned, through performance of Agilent 244K whole genome ChiP-chip analyses of key histone modifications for verified and candidate genes with promoter, CpG island, DNA hypermethylation in the HCT 116 line of human colon cancer cells. The top panel shows, for these cells, average results for the 7.0 kB proximal promoter regions for > 4,500 active genes with non-DNA methylated, CpG island–containing promoters. Note the enrichment of the active H3K4me2 mark on either side of the transcription start sites and the very low levels of the PcG mediated H3K27me3 mark. The middle panels shows the average results for the marks in the same regions of over 40 verified genes with full silencing, and heavily DNA methylated, CpG island promoters (Schuebel et al., 2007; McGarvey et al., 2008). The exact pattern was found for over 6oo such candidate DNA hypermethylated genes in the HCT 116 cells revealed by an expression array discovery approach (Schuebel et al., 2007; McGarvey et al., 2008). Note the marked reduction in levels of, and different positioning of, the active H3K4me2 mark, and a low but distinct level of the H3K27me3 mark broadly distributed over the proximal promoter regions. In the bottom panel, the analysis of the same genes shown in the middle panel have benn performed in an isogenic line of HCT 116 cells, DKO, in which two DNMT’s, DNMT1 and 3b, have been genetically deleted. This maneuver effectively eliminates most of the overall DNA methylation in the cells including in the promoters of the genes analyzed (Rhee et al., 2002). Note the persistence, and even some increase in the H3K27me3 mark. Simultaneously, there is enrichment and re-positioning of the H3K4me2 mark towards the status seen in the active genes in the top panel. This simultaneous pattern for the two marks in the DKO cells, as circled and labeled, resembles bivalent chromatin as per the text.
From all of the above data, and those of others (Cedar and Bergman, 2009), a working model emerges for how a key group of genes with non-DNA methylated, CpG island containing promoters in embryonic cells, may become vulnerable to adoption of abnormal promoter DNA methylation, and attendant tight, heritable silencing, in cancers. In normal settings for cell renewal in adult cell systems, such genes may exhibit bivalent chromatin in stem/progenitor cells but, with signal transduction events attendant to cell maturation, the genes can become active and emphasize the histone modification marks of expressed genes. However, during cancer risk states, such as chronic inflammation, there is abnormal pressure for cell renewal with expansion of stem/progenitor cell pools attempting to provide repair of the cell system. This chronic renewal, in the case of inflammation, occurs in toxic settings wherein cellular increases in, and exposure to, reactive oxygen species (ROS), and other agents (Nelson et al., 2004; Coussens and Werb, 2002; Meng and Riordan, 2006; Nagata, 2005) which can cause DNA damage and other cell injuries, challenge their survival. Abnormal expansion must, then, involve cells adopting survival mechanisms and the maintenance of stem/ progenitor cell characteristics favors this. In turn, the maintenance of these precursor cell states would be favored by retention of a low expression, and the presence of promoter of bivalent chromatin, of many of the genes under discussion. The presence of PcG complexes at these gene promoters may then, as indicated by a body of emerging data (McGarvey et al., 2008; Vire et al., 2006), lead to recruitment of additional silencing protein complexes, including DNA methyltransferases. This would initiate a process of molecular progression and progressive DNA methylation of the gene promoter, CpG islands. In turn, this would favor abnormal cell expansion states which are precursors for tumor progression during which further molecular progression of the abnormal DNA methylation may evolve. These cells would be those, as discussed earlier in this chapter, which can favor further selection of cells which respond to oncogenic gene mutations with growth and survival – and can then help foster progression to invasive states and cancer.
In thinking about why addition of DNA methylation to bivalent chromatin might provide for a more advantageous state of silencing to groups of genes in cancer, several points about this DNA modification must be considered. One can first consider the role of promoter, CpG island DNA methylation in X-chromosome inactivation in females. In this process, silencing of involved genes precedes DNA methylation and is established by a molecular progression initiated by RNA species and recruitment of PcG and other silencing proteins (Brinkman et al., 2006; Schoeftner et al., 2006). However, full maintenance of tight heritable silencing of the affected genes, with little chance for even low level expression and/or gene reactivation, requires the subsequent addition of the DNA methylation (Heard, 2004; Csankovszki et al., 2001).
Our recent studies of differences in higher order chromatin structure for a single gene, GATA-4, which frequently undergoes loss of function associated with promoter DNA methylation in colon and other cancers (Akiyama et al., 2003) suggests a similar function for this DNA modification in tumor progression. This gene encodes a transcription factor important for development of the normal gut endoderm during development, and for proper conversion of proliferative, immature to mature, epithelial cells in adult colon (Kuo et al., 1997; Gao et al., 1998). In normal ES cells, and embryonic carcinoma (EC) cells, GATA-4 is a gene typical for those maintained in a poised, low, expression state associated with bivalent chromatin surrounding the promoter region (Ohm et al., 2007; Bernstein et al., 2006; Mikkelsen et al., 2008; Tiwari et al., 2008). When such cells are induced, with retinoic acid, towards a committed neural state, GATA-4 transcription is increased some 70 to 100-fold and the chromatin around the gene start site adopts an active pattern with loss of the PcG mark, H3K27me3, and gain of the Trx, active mark, H3K4me2 (Tiwari et al., 2008). Comparison of aspects of chromatin around this gene in the above settings versus that for the gene when it is DNA hypermethylated in cultured colon cancer cells reveals much about the way PcG maintains low expression of genes with and without the addition of DNA methylation. First, when the gene is DNA hypermethylated in cancer cells, there is a virtual complete lack of transcription as compared to low, but continued, expression in the EC cells where the gene is marked by PcG constituents and bivalent chromatin (Tiwari et al., 2008). Second, while the PcG, H3K27me3, mark is associated with the GATA-4 gene promoter in this DNA methylated state, levels are lower throughout the region than in EC cells (Tiwari et al., 2008). Third, PcG seems to function for suppressing GATA-4 expression in EC cells by contributing to maintenance of a topologically complex, multi-loop conformation formed by multiple internal long-range contact regions extending over a large region from −30kb upstream to ∼60 Kb downstream of the entire gene (Tiwari et al., 2008). The intersections of the loops are enriched for H3K27me3 and multiple protein constituents of PcG complexes. siRNA mediated depletion of EZH2, which catalyzes the H3K27 me3 mark, leads to a significant reduction in the frequency of the long-range associations which parallels the degree of reduction of H3K27me3 enrichment at the loop intersections (Tiwari et al., 2008).
Fourth, and importantly, the loops around the GATA-4 gene when the promoter has bivalent chromatin in EC cells, and low basal transcription, do not preclude phosphorylated, active Pol2 polymerase from being present at the start site (Tiwari et al., 2008). Furthermore, the poised state of the gene is fully indicated by the fact that, with the marked increase in expression of GATA-4 with induction of neural differentiation, the chromatin loops completely dissolve in concert with loss of PcG proteins and decreases in the H3K27me3 marks. No further increase in the Pol2 polymerase at the gene start site is required for this large increase in gene expression (Tiwari et al., 2008).
Finally, these chromatin studies indicate that the key role of promoter DNA hypermethylation in cancer cells is to tighten the transcription of GATA-4 beyond the repression maintained in the bivalent chromatin state. Thus, in the colon cancer cells, the frequency of many of the long-range interaction points are strikingly increased, some new loops are formed especially in the 3’ region of the gene, and the gene now has no basal transcription and a very low level of Pol2 polymerase at the start site (Tiwari et al., 2008). The loops now encompass multiple, abnormally DNA hypermethylated CpG islands surrounding the entire gene. The methyl-cytosine binding protein, MBD2, is localized to these CpG islands including the one surrounding the gene start site which is classically assessed for DNA methylation (Akiyama et al., 2003). Importantly, when DNA methylation is removed in a cell line isogenic to the colon cancer cells, through genetic disruption of DNA methyltransferases, there is only a modest increase in gene transcription, and while MBD2 is lost, bivalent chromatin is maintained, and the frequency of long-range contacts is only partly diminished to now more resemble those in the undifferentiated EC 2 cells (Tiwari et al., 2008). All of these above findings, modeled in Figure 4, again stress the similarities, between a gene DNA hypermethylated in adult cancer cells and the higher order chromatin conformation for the same gene in embryonic stem/precursor cells. PcG associated loops, forming what may be termed a “repressive hub”, in these latter cells are associated with a normally low, but poised, transcription state. This state can be converted to an active state, with dissolution of the loops, with signal transduction induced cell differentiation. In contrast, addition of DNA methylation in adult cancer cells completely and heritably, represses transcription eliminating the poised state and rendering the gene inaccessible for the transcription that should accompany and help facilitate conversion to a differentiated state (see Figure 4).
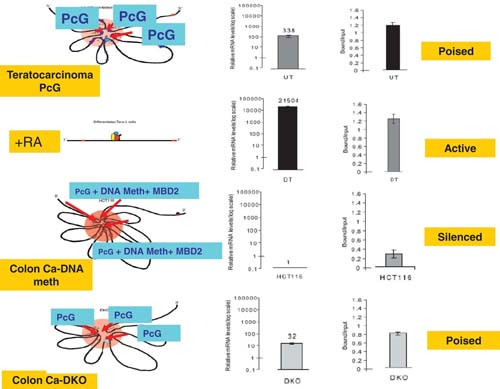
The model is based, as outlined in the text, on experimental studies of the GATA4 gene, frequently DNA hypermethylated early in colon and other cancers (Akiyama et al., 2003). In the top panel (Poised), the gene is in a low poised state in undifferentiated (UT) teratocarcinoma cells as is characteristic in ESC. Characteristics are a broad region of PcG occupancy around the entire gene mediating intersecting loops which intersect the transcription start site. A series of CpG islands in the vicinity of the gene, and including one (blue oval) surrounding the transcription start site are not DNA methylated and phosphorylated PolII is present and poised at the start site. In the second panel (Active), retinoic acid (+RA) has been added to the tetratocarcinoma cells, neural differentiation (DT) has been induced, the gene is markedly activated for transcription, without the need for more PolII recruitment, and this is accompanied by dissolution of the PcG mediated loops. Colored ovals depict active transcription complexes at the gene start site. In the third panel (Silenced), the gene is depicted in HCT 116 colon cancer cells where CpG islands at the gene start site and surrounding the gene (red ovals) are DNA methylated (DNA meth) and encompassed in very tight loops which have PcG still present, but at lower levels, and MBD2 is present at all of the DNA methylated CpG islands. There is no transcription, and PolII at the start site is low. In the bottom panel (Poised), the transcription of the gene has been restored to a poised state in isogenic HCT116 cells where DNA methyltransferases have been genetically deleted (DKO cells (Rhee et al., 2002)) and DNA methylation is absent from the CpG islands. The repression loops are retained, however with less intensity than in wild type HCT116 cells, with somewhat higher PcG occupancy and MBD2 is no longer present.
5. Future directions
The data and working hypotheses discussed in this chapter link embryonic-like chromatin regulation to a molecular progression towards abnormal promoter DNA methylation in cancer cells, and to a role for this latter change, and emergence of embryonic-like cells, in abnormal proliferation in cancer risk states. Certainly, we believe the links are compelling enough to help guide the framework of future research work to validate or refute the hypotheses presented – and, in so doing, such studies may have enormous potential for enriching our understanding of the biology of cancer initiation. Recently, other investigators have emphasized that the gene expression profile of aggressive forms of breast, and other cancers, resembles that of ES cells with increases in PcG gene expression a particularly striking component of the signature (Ben-Porath et al., 2008). This signature applies to tumors such as breast cancers with the state of so called epithelial to mesenchymal conversion, or EMT, which is now a topic of much investigation (Ben-Porath et al., 2008). We believe that the epigenetic abnormalities discussed in this chapter may not only reflect such neoplastic cellular states but also may also may be a key molecular force for their origins and for helping to drive the earliest stages of cancer initiation.
While the above concepts may provide a valuable, and valid, working model to understand the events which initiate abnormal promoter, DNA methylation of large groups of genes in cancer, much remains to be done to follow the model to its ultimate conclusions. The precise molecular events, their temporal occurrences, and the protein complexes involved, must be identified which attract the DNA methylation machinery, and activate its catalytic activity, during abnormal, cellular expansion and tumor initiation. Particularly perplexing is why cells such as ES, contain high levels of multiple PcG proteins and DNMT’s and yet do not DNA hypermethylate genes with bivalently marked promoter, CpG islands. In terms of the key protein complexes, key hints are emerging which link lowering, by histone demethylases, of the active H3K4me marks in bivalent chromatin to the recruitment of DNA methyltransferases, or DNMT’s, to DNA. This recruitment appears to involve complexing of the demethylases to PcG complexes and DNMT's (Pasini et al., 2008; Wang et al., 2009). In turn, lowering the level of H3K4 methylation seems critical for docking of DNMT complexes to DNA in germ cells (Ooi et al., 2007), and inhibition of such a histone demethylase can elicit re-expression of some DNA hypermethylated genes in cancer cells (Huang et al., 2007). Moreover, complexes, again involving direct, or indirect, interaction between PRC2 constituent proteins and DNMT’s, are being suggested (Cedar and Bergman, 2009; Vire et al., 2006), but their nature needs much more clarification.
We have recently presented two experimental models consistent with hypotheses linking stress induced cellular expansion and survival with conversion of genes promoters normally marked by bivalent chromatin to occupation by abnormal DNA methylation. In the first, a double strand break is introduced, in breast cancer cells, into the CpG island of an exogenous promoter construct of the E-cadherin gene which is frequently DNA hypermethylated in this and other cancer cell types. This break leads to rapid recruitment of DNMT's and PRC2 proteins (O’Hagan et al., 2008). While the break is accurately repaired, and the promoter remains functional in most cells, occasional clones retain a silenced promoter which is occupied by DNMT's and which evolves progressive DNA methylation with cell passaging (O’Hagan et al., 2008). In the second model, the CBX7 protein, a member of the PRC1 complex of PcG, is introduced into human teratocarcinoma cells (Mohammad et al., 2009). This results, again with cell passaging, in recruitment of this protein, plus other PcG constituents and DNMT’s, to the promoters of multiple genes which have bivalent marking of their CpG island promoters and progressive evolution of DNA methylation (Mohammad et al., 2009). There is also evidence of direct or indirect interaction between CBX-7 and multiple DNMT's in these cells (Mohammad et al., 2009). Future work in such systems, and those which even more represent early cell transformation stages, will be required to more fully define the nature of the protein complexes involved, their temporal evolution, and their precise role in recruiting the DNA methylation.