
Stem cells of both embryonic and adult origins hold great promise in regenerative medicine owing to their unique properties of unlimited self renewal and differentiation toward specific lineage(s) once they receive the proper signals. Proteomics is a series of technology platforms driven by advancements in mass spectrometry and bioinformatics that encompass protein identification, the relative quantitation of proteins and peptides, their subcellular localization, and studies of post-translational modifications and protein-protein interactions. Stem cell biology has been influenced by these approaches and has evolved in the post-genomics era. Among many challenges in stem cell biology, there is a pressing need for the implementation of proteomic applications. Recent work on stem cells using proteomics has shown that transcriptome analyses fail to provide a full guide to developmental change in stem cells, and protein interactions that can only be discovered systematically using proteomic approaches have yielded important new concepts on processes regulating development and stem cell pluripotency. In this chapter, we will review current proteomic studies on embryonic and adult stem cells with an emphasis on embryonic stem cells.
1. Introduction
1.1. Stem cells and proteomics
Stem cells of any type are defined by two distinct properties. The first is indefinite self-renewal, a feature that provides for maintenance in a tissue and/or organism for an extended period of time. The second is the ability to differentiate into a number of different daughter cell types, unlike non-stem cells that are committed to a single lineage. Adult somatic stem cells are found in the majority of organs and tissues in adult organisms, and are thought to function in long-term tissue maintenance and/or repair. In contrast, embryonic stem cells (ESCs) are derived from embryos and are unique in their ability to be maintained in vitro in a pluripotent state, i.e., capable of recapitulating all three germ layers and an entire organism.
Both adult and embryonic stem cells have provided distinct challenges for analysis. Adult stem cells tend to be rare, difficult to purify or maintain in culture. For this reason, adult stem cells have provided a greater technical challenge for large-scale transcriptome and proteome analyses. ESCs, by contrast, are readily grown to large numbers in culture, and have been utilized for extensive analysis by transcriptional profiling and other genome wide techniques. In addition, ESCs are easily manipulated in vitro, making them ideal tools for probing stem cells properties and characteristics using a wide variety of techniques. In the current post-genomic era, in which transcriptome mapping using DNA microarray technology is commonplace (Ivanova et al., 2002; Ramalho-Santos et al., 2002), consideration of the transcriptome alone offers an incomplete and biased interpretation of the underlying stem cell biology (Evsikov and Solter, 2003: Fortunel et al., 2003). Inherent problems associated with such a transcriptional profiling approach include first, the analysis is obviously limited to genes present on the microarray, and it is possible that there are "stemness" genes that have not yet been identified and are not represented in the chips used; second, changes at the mRNA level may not be proportional to changes in protein expression (Gygi et al., 1999a); third, protein complex formation, numerous post-translational modifications (PTMs) and protein degradation greatly impact protein-protein and protein-DNA interactions, making the functional output of these systems virtually impossible to predict based solely upon gene expression and/or genomic data.
The term “proteome” was originally coined by Wilkins et al. (Wilkins et al., 1996) to describe the total set of proteins expressed in a given population, a.k.a. cell, tissue, organelle, organism, or pathological state. The term “proteomics” refers to a set of techniques well suited to identify proteomes, but has been broadened to include large-scale techniques capable of identifying proteins, and analyzing both their structures and their functions at a genome wide level. Proteomics encompasses a wide variety of techniques, ranging from yeast two-hybrid screens for identifying protein-protein interactions (Rual et al., 2005), antibody-based protein chips for identifying proteins (MacBeath, 2002), and high throughput crystallography screens (Stevens et al., 2001) to provide structural analysis. All of these techniques (see summary in Figure 1) provide invaluable insights into the proteome and its function in a cell, but perhaps the most widely utilized group of techniques center around mass spectrometry (MS), which will be further discussed below (also reviewed in Aebersold and Mann (2003) and Cravatt et al. (2007)).
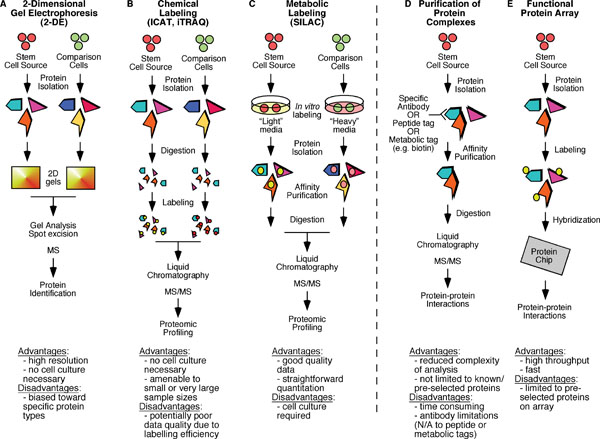
Advantages and disadvantages of the various methods that have been used in proteomic profiling and protein interactome mapping are highlighted. A. Two dimensional electrophoresis (2-DE) followed by mass spectrometry (MS) analysis has been widely used to compare various populations of stem cells to more differentiated cell types. B. iTRAQ (isobaric tags for relative and absolute quantification) and ICAT (isotope coded affinity tags) are two chemical labeling approaches that have been used prior to tandem mass spectrometry (MS/MS) analysis in comparison of purified stem cell populations. C. SILAC (stable isotope labeling with amino acids in culture) is a metabolic labeling approach that has been used in proteomic profiling and quantitative phosphoproteomic studies. D. Affinity-based purification of protein complexes followed by MS/MS analysis has been widely used in protein interactome studies. E. The use of functional protein arrays is a very promising approach that has predominantly been utilized in yeast protein interactome studies.
1.2. Mass spectrometry (MS)-based platforms for proteomic research
MS functions by ionizing relatively small molecules and then measuring their mass to charge ratio (m/z). While traditional MS itself is capable of identifying the mass of a highly purified small molecules, it can do little else with more complicated molecules (such as peptides) or mixtures of samples. To further increase the range of substances that can be identified by MS, two can be combined in tandem (termed MS/MS) in which a peptide first has its molecular mass measured (in MS1) and then is bombarded with electro-neutral gases to cause fragmentation. The m/z ratios of these resulting smaller fragments are then measured in the second analyzer (MS2), and following computer analysis the amino acid sequence of the peptide can be determined (see Figure 2). Thus, virtually any single peptide, in a relatively purified state can be identified using tandem MS. For the analysis of a whole protein, or even multiple proteins, further manipulations are needed to purify the mixture to reduce the complexity of any specific input into the MS. This is usually accomplished first by purifying a sample by simple SDS-PAGE electrophoresis and subsequent excision of relevant band(s) or a whole lane of a gel which is then broken down into smaller fragments. These gel fragments are then digested in situ with a protease (typically trypsin) and the peptides are recovered. To further fractionate the specimen prior to analysis by MS, the samples undergo either single dimensional liquid chromatography (LC, typically a reverse-phase LC which separates based upon hydrophobicity), or multidimensional (LC/LC), with the choice based upon the complexity of the initial sample. Subsequent to LC or LC/LC but prior to application to the mass analyzer, the peptides are ionized, usually by electrospray ionization in which a potential is applied across a fine needle through which passes the elute from the LC column, creating a fine spray that forms droplets containing the sample, and heat applied prior to entry into the MS allows for desolvation and ionization. MS/MS analysis ensures identification of the peptide size and amino acid sequence.
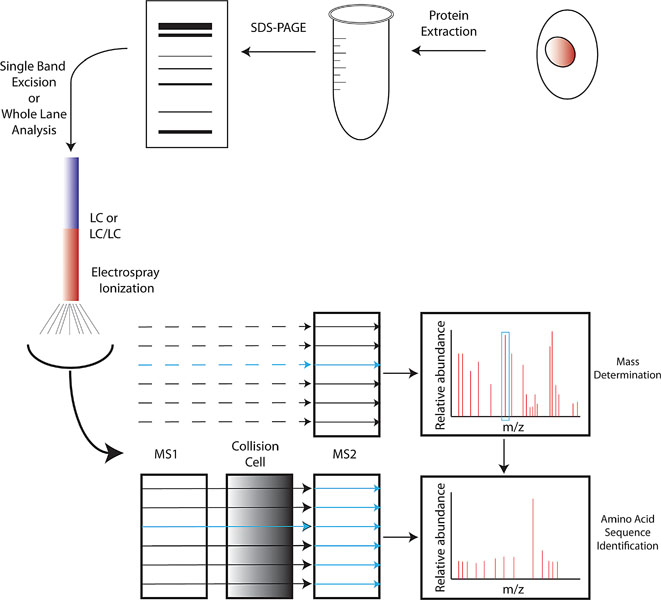
Protein extracts are made from stem cells of any type and first fractionated by SDS-PAGE. Individual bands or the whole lane are then subjected to in situ digestion with trypsin. After trypsin digestion, the product is subjected to either single-dimensional (LC) or multi-dimensional (LC/LC) liquid chromatography for further separation of the mixture. Elute from LC is then ionized by electrospray ionization, and each elute peak first passes through an MS (typically MS2) for mass determination. Each peak is then separated in MS1 and passes into a collision chamber where it is further fragmented and subsequently analyzed in MS2, which aids in peptide sequence determination.
After completion of this process, a complicated protein mixture is reduced to a fragment ion spectrum and molecular weight for each peptide. Bioinformatics is then necessary to translate each specific spectrum into a peptide and protein from which it originally arose. The algorithms involved are varied and complex, but are based upon comparison to the theoretical spectrum of known proteins from a database and de novo sequencing in which each fragment spectrum is directly translated into a specific peptide, or a hybrid approach that combines both (reviewed in (Nesvizhskii et al., 2007). Each peptide is then mapped to a protein based upon either a deterministic (i.e., a predetermined algorithm such as in (Resing et al., 2004: Tabb et al., 2002) or probabilities of a match (Price et al., 2007). The result is an identification of all possible protein(s) in a given sample.
The use of LC coupled tandem MS/MS has allowed for two general approaches. The first is termed “shot-gun” proteomics, in which a single sample, such as a cell line, tissue, or highly purified cell population is analyzed to assess all peptides/proteins expressed. This is also known as expression-based proteomics. The second is affinity purification, in which a single protein species is purified from a cell; with the goal being to identify associated molecules (see Figure 1D). While both methods have been widely utilized, affinity purification has provided unique insights into network properties of organisms (Gavin et al., 2002) and stem cells (see Section 2), and thus often been referred to as functional proteomics (Kocher and Superti-Furga, 2007). In general, affinity purification is based upon two techniques. First, affinity purification can be performed on native proteins using antibodies to isolate a single protein and its associated proteins (Uhlen and Ponten, 2005). The major drawback is that the antibody can often be the limiting reagent, making it difficult to purify rare proteins or large amount of complexes. The second involves attaching a specific peptide tag to a cDNA of interest, allowing for easy purification and elution of the tagged protein of interest (Rigaut et al., 1999). These methods also typically allow for eluting the affinity tagged complexes from the column by proteolytic cleavage at a specific recognition sequence (e.g., TAP tag in Figure 3A). A variation of this tag-based technique is based upon metabolic tagging with biotin (de Boer et al., 2003). Cells are generated which express the E.Coli derived BirA ligase capable of attaching biotin to a specific peptide recognition sequence. cDNAs are then engineered to contain the recognition sequence, allowing them to be efficiently biotinylated in vivo and captured in vitro due to the strong affinity of biotin for streptavidin (see Figure 3B). The predominate advantage of metabolic tagging methods is the exceptionally high affinity of streptavidin for biotin (Kd ≈ 10−15, as opposed to a Kd ≈ 10-9 for calmodulin binding protein). Using either tagging approach, the tags are often combined to allow for tandem purification, thereby increasing the purity of the complex and the specificity of the subsequently identified interactions. There are several advantages associated with this affinity purification-MS method: first, it can be performed under relatively physiological conditions; second, it does not typically perturb relevant PTMs, which are often crucial for the organization and/or activity of complexes and can also be identified by MS; third, it can be used to probe dynamic changes in the composition of protein complexes when used in combination with quantitative proteomics techniques such as iTRAQ and SILAC (see below).
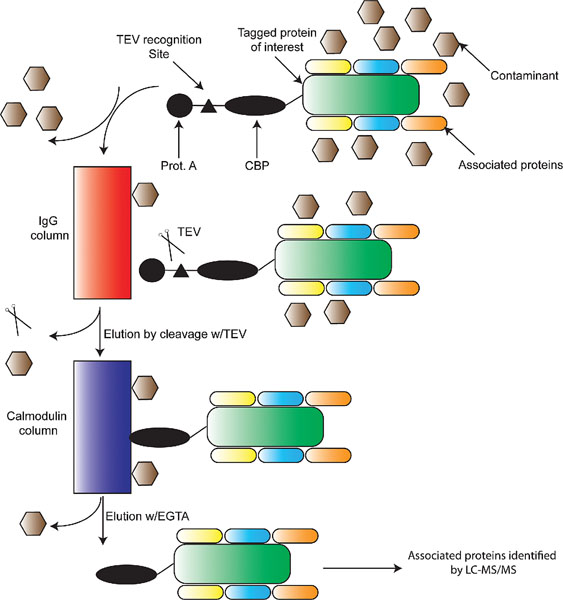
A. Tandem affinity chromatography: a protein of interest is first engineered to contain a Protein A (filled in circle) tag, a Tobacco Echo Virus recognition site (TEV, filled in triangle), and a calmodulin binding peptide (CBP) tag (filled in ellipse). Extracts are made from cells expressing the tagged protein, which should contain associated proteins and contaminants. These complexes are bound to an IgG column, and washed to remove majority of contaminants. TEV protease is then used to elute the semi-purified protein complexes which are subsequently absorbed onto a calmodulin column. After further washing, purified protein complexes containing the protein of interest and its associated proteins are eluted by calcium chelation (EGTA) and identified using LC-MS/MS.
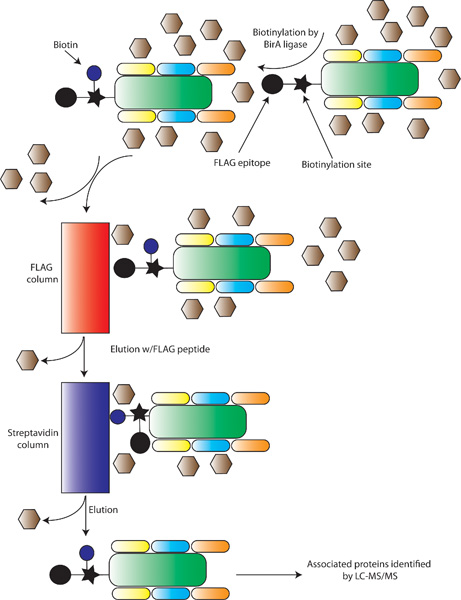
B. Metabolic tagging for affinity chromatography: First, stem cells are engineered to express the E.Coli derived biotin-ligase BirA, which attaches biotin to a defined recognition sequence, shown with a filled-in star. Proteins of interest are then engineered with a biotinylation site and a FLAG epitope (shown as a filled-in circle). Inside the stem cell, the biotin (blue circle) is added by BirA. Protein extracts are made and applied to a FLAG-antibody column, and after washing semi-purified complexes are eluted with FLAG peptide. The elute is then applied to a streptavidin column, and after washing the purified protein of interest and its associated proteins are eluted by denaturing and identified by LC-MS/MS.
In addition to identifying large arrays of proteins as well as protein complexes, proteomics has also advanced to be more quantitative, i.e., allow for protein levels to be directly compared between two samples (Oda et al., 1999: Ong et al., 2003). While there are a number of techniques (Summarized in Figure 1A–C), two are most widely used: ICAT (isotope coded affinity tags; Gygi et al. 1999b) and iTRAQ (isobaric tags for relative and absolute quantification) (Ross et al., 2004). Briefly, proteins from two populations of cells are labeled using different chemicals with different isotope compositions (i.e., hydrogen vs. deuterium in the case of ICAT or an analogous four isotope tag in iTRAQ), and the samples are then remixed and quantitative protein levels can be assessed. The advantages of these techniques are that both allow for the quantitation of virtually any sample, and very large samples are possible, although issues with labeling efficiency and over-labeling can cause difficulties. In contrast, SILAC (stable isotope labeling with amino acids in culture; Chen et al. 2000: Ong et al., 2002: Zhu et al., 2002) uses a similar approach in which two populations of cells are labeled with isotopically distinct amino acids in vivo and then analyzed, allowing for differences between the two cell populations to be assessed. The advantage of this technique is that labeling efficiency and over-labeling are no longer an issue, although it is a difficult procedure to scale up to larger, proteome scale procedures. The development of these MS-based technique platforms has greatly advanced the proteomic studies of stem cells, which is discussed in detail in the next two sections.
2. Proteomic studies of embryonic stem cells (ESCs)
2.1. The ESC proteome
Since their discovery over 25 years ago (Evans and Kaufman, 1981), murine embryonic stem cells (mESCs) have provided an invaluable tool for answering genetic questions (Thomas and Capecchi, 1987). With the establishment of human embryonic stem cells (hESCs; Thomson et al. 1998), new opportunities for tissue repair or replacement are being actively explored. To complement the transcriptomic analyses of ESCs that define a genome wide RNA expression signature of stemness (Ivanova et al., 2002: Ramalho-Santos et al., 2002), stem cell proteomics provides an excellent tool to characterize ESCs at protein level and derive a protein pluripotency signature that may disclose novel ESC-specific benchmarks.
The proteomic analysis of embryonic stemness has been probed using MS-based protein profiling of both undifferentiated and differentiated ESCs. A quest for human (line HES-2) and mouse (line D3) ESC-specific proteins resulted in 1,775 non-redundant proteins in hESCs, 1,532 in differentiated hESCs, 1,871 in mESCs, and 1,552 in differentiated mESCs with a false positive rate of <0.2%. Comparison of the data sets distinguished 191 proteins exclusively identified in both human and mouse ESCs, many of which are uncharacterized proteins and are potential novel ESC-specific markers or functional proteins (Van Hoof et al., 2006). Elliott et al. utilized 2D gels with multiple pH gradients and varied acrylamide concentrations to resolve approximately 600∼1000 protein spots from mouse R1 ESCs on silver stained gels and represents the initial step in producing a comprehensive ESC 2D protein database (Elliott et al., 2004). Nagano et al. using an automated microscale 2D LC-MS/MS analyzed total proteins in mouse E14-1 ESCs (Nagano et al., 2005). They assembled a catalogue consisting of ∼1800 proteins, containing many components derived from ESC-specific and stemness genes defined by the transcriptome analysis (Ramalho-Santos et al., 2002), and a number of components, such as Oct4 and UTF1, which are expressed specifically in ESCs. Importantly, they detected ESC-specific transcription factors of low abundance (104 to 105 copies/cell) and found 36% of total proteins were located in the nucleus, consistent with the high nuclear to cytoplasmic ratio of ESC colonies.
Recently, Graumann et al. fractionated the SILAC-labeled ESC proteome by 1D/IEF (isoelectric focusing) followed by high resolution analysis on a linear ion trap-orbitrap instrument (LTQ-Orbitrap) to sub-ppm mass accuracy which resulted in confident identification and quantitation of more than 5,000 distinct proteins (Graumann et al., 2007). This is the largest quantified proteome reported to date and contains prominent stem cell markers, such as Oct4, Nanog, Sox2, Utf1 and an embryonic version of Ras (ERas). Bioinformatics analysis of the ESC proteome reveals a broad distribution of cellular functions with overrepresentation of proteins involved in proliferation. In addition, Graumann et al. compared the proteome with a recently published map of chromatin states of promoters in ESCs (Mikkelsen et al., 2007) and find excellent correlation between protein expression and the presence of active and repressive chromatin marks.
An interesting feature of the ESC proteome in the Nagano study (Nagano et al., 2005) and another study in D3 ESCs (Nunomura et al., 2005) is that it retains the cell surface markers and signaling molecules that are characteristic of differentiated cells. This is not inconsistent with the notion that interactions between cell surface proteins and extracellular ligands are key to initiating ESC differentiation to specific lineage. Although it is formerly possible that a small portion of cells were differentiated to a variety of cell lineages during the culturing condition, it is tempting to hypothesize that the ESC proteome is equipped with multiple protein components unique to a number of differentiated cell types, enabling cells to respond to various external signals leading to differentiation to specific lineages, a property of pluripotency of the ESCs. So far, relatively little is understood regarding how stem cells are programmed toward a particular cell lineage. This is an important area of investigation that involves directed differentiation to influence the lineage commitment of these pluripotent cells in vitro. Manipulation of extracellular signals and overexpression of transcription factors can drive ESCs to commit to a specific cell type, however, ultimately it is the changes in nuclear expression that direct differentiation down to a specific lineage. Accordingly, nuclear proteomics–studies of collective actions and interactions of proteins found in the nucleus–has been proposed (Barthelery et al., 2007) to inventory nuclear proteins in both undifferentiated and differentiating cells and decipher their dynamics during cellular phenotypic commitment. This provides an opportunity to identify unknown transcription factors and additional nuclear effectors critical in the maintenance of cellular phenotype. In addition, it offers insights as to what nuclear profile is needed to program or reprogram cellular fate with limited imprinting side effects (Barthelery et al., 2007).
2.2. The ESC epiproteome
To identify biologically relevant proteins important for stem cell self renewal and pluripotency, the extensive catalogue and benchmark of protein databases are not sufficient. Many biochemical pathways are directed by changes in PTMs such as phosphorylation rather than by changes in abundance of proteins themselves. Studies have now shown that epigenetic mechanisms, such as covalent modifications of histones and DNA methylation are vitally important to the pluripotent nature of ESCs and that these mechanisms also regulate differentiation (Atkinson and Armstrong, 2008). The epigenetic nature of the ESCs (the ESC “epigenome”) has been demonstrated to be unique and its characteristics have been strongly linked to the global permissivity of gene expression and pluripotency (Niwa, 2007). In analogy to epigenome, a new term “epiproteome” has been coined to reflect a protein landscape of PTMs and histone variants (Dai and Rasmussen, 2007).
Phosphorylation is a critical PTM involved in modulating protein function. To gain insight into intracellular signals governing ESC self-renewal and differentiation, a multivariate systems analysis of proteomic data generated from combinatorial stimulation of mESCs (line CCE) by fibronectin, laminin, LIF and fgf4 was performed (Prudhomme et al., 2004). Phosphorylation states of 31 intracellular signaling network components were obtained across 16 different stimulus conditions at three time points by quantitative Western blotting, and computer modeling was used to determine which components were most strongly correlated with cell proliferation and differentiation rate constants obtained from measurements of Oct4 expression levels. The study identified a set of signaling network components most critically associated with differentiation, proliferation of undifferentiated as well as differentiated cells.
A large-scale proteomic analysis of hESCs (BG01 and BG03 lines) was also performed using PowerBlot and Kinexus Western blot assays coupled with immunofluorescence (Schulz et al., 2007). The study identified over 600 proteins expressed in undifferentiated hESCs, including a number of potential new stem cell markers, and highlighted over 40 potential protein isoforms and/or PTMs including 22 phosphorylation events in cell signaling molecules. More recently, a nucleosome-ELISA method was developed to assess quantitatively the status of PTMs and histone variants (dubbed “epiproteomic signature”) present within the total cellular nucleosome pool (Dai and Rasmussen, 2007). The results indicate that assessment of the steady-state levels of PTMs and macroH2A yields an epiproteomic signature that can distinguish between ESCs, EC cells and MEFs. Furthermore, epiproteomic nucleosome signatures change in response to exposure of cells to small molecules such as RA and TSA and over the course of ESC differentiation. This indicates that the epiproteomic signatures are useful for investigation of stem cell differentiation, chromatin function, cellular identity and epigenetic responses to pharmacologic agents.
The direct analysis of a large number of peptides using 2D LC-MS/MS permitted the systematic identification of peptides carrying PTMs (Witze et al., 2007). Nagano et al. identified protein PTMs in a number of ESC proteins including five Lys acetylation sites and a single phosphorylation site (Nagano et al., 2005). Phosphorproteome analysis of undifferentiated and differentiated mESCs (line J1) using phosphoprotein affinity purification followed by 2D LC-MS/MS indicated that many chromatin-remodeling proteins are potentially regulated by phosphorylation (Puente et al., 2006). Interestingly, affymetrix microarray analysis indicated that gene expression levels of these sample proteins had minimal variability between the compared samples (Puente et al., 2006). These findings collectively highlight the critical roles that epigenetic factors play in maintaining pluripotency of ESCs (Bibikova et al., 2008), and stress the necessity and value of proteomic analysis.
2.3. The ESC protein interaction network
The expression-based studies of ESC proteome and epiproteome provide a comprehensive inventory of proteins as well as their PTMs, some of which may be used as ESC markers. However, such protein lists are not sufficient to describe biological processes. Vital cellular functions require the coordinated action of a large number of proteins that are assembled into an array of multiprotein complexes of distinct composition and structure. The analysis of protein complexes and intricate protein-protein interaction networks is a key to understanding virtually any complex biological systems including stem cells (Levchenko, 2005).
To understand how pluripotency is programmed and maintained in ESCs, we have utilized a proteomic approach to isolate protein complexes and constructed a protein interaction network surrounding the pluripotency factor Nanog (Wang et al., 2006). The approach takes advantage of the extraordinary affinity of streptavidin for biotin, and obviates reliance on antibodies of inherently lower affinity for purification (see Figure 3B). It has been reported that single-step streptavidin capture of tagged transcription factors is sufficient to isolate specifically associated proteins with minimal non-specific contamination (de Boer et al., 2003). In this system, BirA expressing ESCs serve as a recipient for other tagged cDNAs. A construct bearing the pluripotency factor with a FLAG tag as well as a peptide tag that serves as a substrate for in vivo biotinylation was expressed in ESCs (see Figure 4A). The tagged protein was recovered from nuclear extracts with streptavidin beads together with its potential interacting partners. For tandem purification, the nuclear extracts were first subjected to immunoprecipitation with anti-FLAG antibodies and the recovered protein complexes were further purified by streptavidin beads. Protein complexes recovered from either one-step streptavidin or tandem purification were subjected to microsequencing by LC-MS/MS (see Figure 4B).
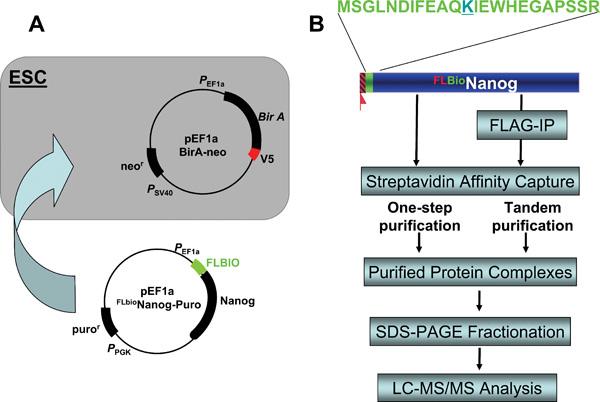
A. Establishment of a biotinylation system in ESCs. A stable ESC line expressing the bacterial BirA enzyme was first established by transfection with a BirA-expressing plasmid bearing the neomycin resistance (neor) gene and G418 selection; A second plasmid containing Nanog cDNA with an N-terminal Flag-biotin dual tag (FLBIO) and a puromycin resistance (puror) gene was introduced and cells selected with puromycin. The resulting stable lines are resistant to both G418 and puromycin and express FLAG-tagged, biotinylated Nanog that can be immunoprecipitated by anti-FLAG and streptavidin antibodies/beads. B. Two complementary affinity purification strategies for protein compexes purification. Single streptavidin immunoprecipitation and tandem affinity purification (anti-Flag immunoprecipitation followed by streptavidin pulldown) were performed in parallel, the purified protein complexes were fractionated on SDS-PAGE, and subjected to LC-MS/MS to identify components of the protein complexes.
We first chose to focus on the variant homeobox Nanog protein, considering its role in maintaining pluripotent state of cells in early mouse embryo and promoting pluripotency of mESCs (Chambers et al., 2003: Mitsui et al., 2003). By affinity purification of Nanog associated protein complexes followed by LC-MS/MS, components of Nanog protein complexes (and thus direct and/or indirect Nanog-interacting partners) were identified. Many of the candidates identified were other transcription factors or components of transcriptional complexes, some of which had already been associated with ESC functions in previous studies. A number of novel (e.g., Dax1, Rif1, Nac1 and Zfp281) and known (e.g., Oct4) critical factors were validated, both physically and functionally, for association with the bait Nanog and were used (together with another well known ESC marker Rex1) for purification of a second tier of complexes. The resulting datasets were used to generate a complex network of interacting proteins that is concisely depicted in Figure 5A. This iterative, “bottom-up” strategy reveals a tight, highly interconnected protein network greatly enriched in nuclear factors individually required for maintenance of ESC properties and co-regulated on ESC differentiation (Wang et al., 2006). In addition, the network links to multiple corepressor pathways, which provides both a means to regulate different sets of target genes and a fail-safe mechanism to prevent differentiation toward different lineages, a requisite for pluripotency. Furthermore, downstream gene targets of several core pluripotency factors (e.g., Nanog, Oct4) identified from previous studies (Boyer et al., 2005: Loh et al., 2006) also serve as upstream regulators in the network (see Figure 5B), indicating that the ESC interaction network is a self-contained, exceedingly tight cellular module dedicated to pluirpotency. Finally, identification of a number of network proteins that are not strictly specific to ESCs and cannot be identified by transcriptional profiling, highlights the importance and advantage of proteomic studies in ESCs.
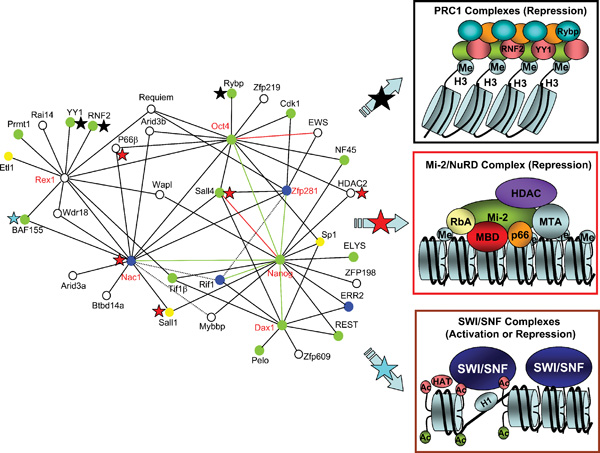
A. Proteins with red labels are tagged baits for affinity purification. Green and red lines indicate confirmed interactions by coimmunoprecipitation or published data. Dotted lines indicate potential association. Green circles indicate proteins whose knockout results in defects in proliferation and/or survival of the inner cell mass or other aspects of early development; Blue circles indicate proteins whose reduction by RNAi (or shRNA) results in defects in self-renewal and/or differentiation of ESCs; Yellow circles are proteins whose knockout results in later developmental defects; White circles denote proteins for which no loss-of-function data are available. Also indicated within the network are three major chromatin modifying complexes whose components are marked with black stars (Polycomb repression complex 1), red stars (NuRD complex) and a blue star (SWI/SNF complex), respectively.
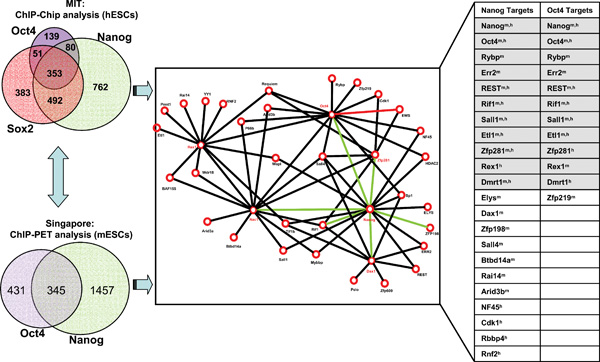
B. Targets of pluripotency factors are highly represented in the network. Left panels show the targets of Nanog, Oct4 and Sox2 in hESCs (Boyer et al., 2005) and targets of Nanog and Oct4 in mESCs (Loh et al., 2006). The right table summarizes the targets of Nanog and Oct4 from the two ChIP studies (left) that are present in the protein network (middle). Note: Xm, h indicates that gene X identified as targets of Nanog and/or Oct4 in both mouse (m) and human (h) ESCs. Shaded are the targets of both Nanog and Oct4.
The ultimate goal of functional proteomics in stem cells is to decipher the molecular function of an entire cell by generating a construction master plan describing all molecular machines, their functions in maintain stem cell properties, their reactions to external stimuli during differentiation, and their interconnectivities. Our work on the protein interaction network in mESCs described above represents the first step toward that direction. In addition, it provides a framework for exploring the combinations of factors that may permit optimal reprogramming of differentiated cells to an ES cell state (Wang and Orkin, 2008).
2.4. The ESC transcriptional regulatory network
Large-scale transcriptomic and proteomic analyses of ESCs are complementary to each other and have laid a foundation for a better understanding of the underlying stem cell biology. However, missing links exist such as gene transcription may not directly be indicative of or proportional to protein translational readout (expression), and conversely, protein expression and multiprotein complexes do not themselves specify target gene regulation of the protein(s). A comprehensive understanding of establishment of the pluripotent state in ESCs requires construction of an expanded transcriptional regulatory network in which many key transcription factors besides Nanog, Oct4 and Sox2 and their interaction partners (Wang et al., 2006) bind directly to their target genes.
Recent studies have begun to elucidate transcription networks surrounding the three core ESC transcriptional factors Nanog, Oct4, and Sox2 that operate to control ESC pluripotency. Using ChIP-chip analysis (chromatin immunoprecipitation followed by microarray hybridization to identify binding sites on a genome wide scale), Boyer et al. showed that Oct4, Sox2 and Nanog collaborate to regulate hESC pluripotency and self-renewal through autoregulatory and feedforward loops. These three transcription factors function by activating pluripotency genes including themselves and by repressing key developmental genes possibly in part with aid of Polycomb proteins (Boyer et al., 2006: Lee et al., 2006). Using ChIP followed by paired-end ditags (ChIP-PET) approach, Loh et al. surveyed target genes of Nanog and Oct4 in mESCs and found that both regulate substantially overlapping target genes (Loh et al., 2006). However, cross-examination of the target genes of Nanog and Oct4 between hESCs and mESCs revealed a limited overlap between the two sets of data, suggesting either different control mechanisms between the two species or inherent variations between the two technique platforms. The result emerged from these studies was the high degree of overlap between the genes targeted by pairs or all the three transcription factors. However, questions remained to be address as how other factors besides the three in the protein interaction network (see Figure 5A) contribute to maintenance of stem cell identity and how the multiprotein complexes specify target gene regulation.
Although neither expression nor transcription factor binding studies in isolation are sufficient to establish a regulatory relationship between a transcription factor and its targets, integrating these methodologies has provided two independent sources of evidence for high confidence prediction of novel transcriptional networks regulating ESC self-renewal and commitment (Walker et al., 2007: ). Using a modified ChIP-chip procedure (dubbed bioChIP-chip) combined with affinity purification and LC-MS/MS (dubbed bioSAIP-MS) to expand the current protein interaction network (see Figure 6), Kim et al. systematically surveyed target genes of total 9 protein interaction network factors (Nanog, Oct4, Sox2, Klf4, c-Myc, Nac1, Zfp281, Dax1 and Rex1) and constructed an expanded transcriptional regulatory network in mESCs (Kim et al., 2008). This network contains many more core pluripotency factors in addition to Nanog, Sox2 and Oct4 that form autoregulatory and feedforward regulatory circuitries. In particular, Klf4 serves as an upstream regulator of larger feedforward loops containing Nanog, Sox2 and Oct4 as well as c-Myc. More importantly, combined analyses of bioChIP-chip data with gene expression data revealed that majority of common targets of over 4 factors are highly active in ESCs and repressed upon differentiation. In the case of targets bound by fewer factors, both active and repressed genes are present and the balance shifts toward gene inactivity with reduced factor co-occupancy. The extreme is that distinct targets of a single factor are largely inactive or repressed. Moreover, the regulatory network also indicates that c-Myc and three other factors (Nanog, Oct4, Sox2) play distinct roles in ESCs, i.e., c-Myc is largely involved in stimulation of cell proliferation and regulation of chromosomal accessibility; whereas Oct4/Sox2/Nanog positively regulate ESC factors and negatively regulate differentiation (Kim et al., 2008). This provides a potential mechanism that might account for the differential regulation of transcription factor targets in ESCs and provides mechanistic insights into the 4-factor (Oct4, Klf4, Sox2, and c-Myc) mediated somatic cell reprogramming (Lewitzky and Yamanaka, 2007).
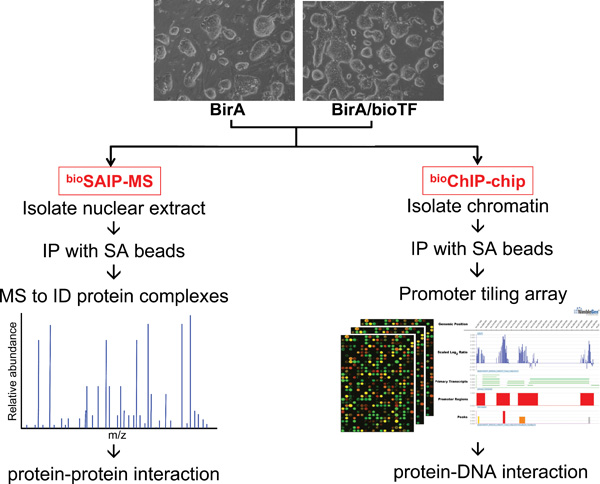
The ESCs expressing BirA alone (as control) and BirA plus biotinylated transcription factors (bioTF) can be used for isolation of protein complexes using streptavidin (SA) immunoprecipitation (IP) coupled with LC-MS/MS (dubbed bioSAIP-MS) and construction of a protein-protein interaction network; meanwhile, the same ESCs can be subjected to in vivo biotinylation-mediated chromatin immunoprecipitation and microarray (dubbed bioChIP-chip) to identify protein-DNA interactions and construct a transcriptional regulatory network.
Our demonstration of in vivo biotinylation of tagged proteins and streptavidin affinity capture to identify global targets of multiple factors involved in the transcriptional control of pluripotency in ESCs further highlights the power of proteomic approaches to define in a systematic fashion the protein-protein interaction and protein-DNA interaction networks operative in ESCs. In particular, affinity purification of biotin-tagged protein complexes coupled with LC-MS/MS (bioSAIP-MS) and the bioChIP-Chip method obviates reliance on low-affinity antibodies and allows for the generation of two independent data-rich resources with the same biotin-tagged cell lines and similar procedures (see Figure 6), paving the way for highly efficient, large-scale proteomic studies in ESCs.
3. Proteomic studies of adult stem cells
3.1. Current status of adult stem cell proteomics
Somatic stem cells have been identified within adult organisms, and are defined by their dual properties of self-renewal and differentiation. Unlike ESCs, however, adult somatic stem cells are restricted in their ability to give rise to cell types within a defined lineage. Over the last 20 years a large body of work has been compiled to further define these cells, develop rigorous isolation strategies, deduce their in vitro and in vivo functions, and establish transcriptional profiles. While these studies have greatly advanced the field, a complete understanding of the mechanisms that regulate self-renewal and potency within adult stem cells requires integration of multiple high-throughput platforms assessing transcriptomes, proteomes and protein interactomes. Unlike ESCs, however, only a relatively small number of studies have ventured into proteomic profiling and protein interaction mapping of adult stem cells. The majority of studies in the field of adult stem cell proteomics have focused on three cell types: hematopoietic stem cells (HSCs), neural stem cells (NSCs) and mesenchymal stem cells (MSCs). There are many inherent challenges in pursuing proteomic studies using adult stem cells. With the exception of NSCs, which can be significantly expanded in vitro without loss of stem cell properties, most adult stem cell types cannot be maintained or expanded in culture without inducing changes in their potency. Thus, there are limits to the numbers of available input cells and unlike the development of global nucleic acid amplification for transcriptional profiling, currently there is lack of an effective protein amplification method.
Many of the initial proteomic efforts from in vitro expanded adult stem cells have utilized 2-dimensional gel electrophoresis (2-DE) as a front-end fractionation method prior to mass spectrometry (MS) analysis (see Figure 1A). There are several limitations inherent to this approach, including limited resolving power, poor representation of very large or small, basic or hydrophobic proteins, the requirement for relatively large amounts of sample, and statistical issues (different analysis algorithms generate divergent results). Combined data sets from these proteomic profiling studies reveal that the largest conserved group of proteins in adult stem cells are involved in energy metabolism (Baharvand et al., 2007). However, these data sets are largely biased by the methodology used and consequently may simply represent the most abundant proteins broadly expressed among these cell types.
Subsequent to these initial studies, several groups have taken advantage of the development of more sophisticated and unbiased proteomic techniques to gain new insights into adult stem cell biology. Development of sensitive iTRAQ methodology combined with MS analysis (see Figure 1B) has allowed comparison of purified populations of hematopoietic stem and progenitor cells with as few as 1x106 input cells. Interestingly, results of this study suggest that HSCs, unlike their more differentiated progenitor counterparts, are adapted for anaerobic environments (Unwin et al., 2006). These differences were not seen when the transcriptomes of these same populations were compared (Unwin et al., 2006), strongly indicating that transcriptional profiling alone would not have been sufficient to deduce this novel aspect of HSC biology. Additionally, iTRAQ has been effective in defining a poorly characterized population of hematopoietic progenitor cells (Lineage- c-Kit+ Sca-1−) as principally erythroid in nature (Spooncer et al., 2007). In the MSC field, 2-dimensional liquid chromatography (2D LC) or LC/LC fractionation followed by tandem MS/MS has been utilized to demonstrate that osteogenic differentiation of stem cells results from the focusing of gene expression in functional clusters rather than simply from the induced expression of new genes (Salasznyk et al., 2005).
It has also been demonstrated that PTMs can significantly influence adult stem cell fate decisions. A quantitative phosphoproteomics approach, facilitated by SILAC technology (see Figure 1C), has been used to study the influence of growth factor signaling on MSC differentiation. Specifically, the mechanism by which two related growth factors (EGF and PDGF) differentially impacted MSC differentiation was found to be mediated by tyrosine phosphorylation (Kratchmarova et al., 2005).
As highlighted earlier in this review, the recent characterization of a functional protein interactome and transcription regulatory network in mESCs (Kim et al., 2008: Wang et al., 2006) has yielded important new concepts in processes regulating development and stem cell pluripotency. While this type of intricate network has not yet been identified within adult stem cells, initial efforts towards this goal have utilized a proteomics approach to identify critical protein-protein interactions regulating self-renewal and differentiation. Using antibody-based purification of protein complexes (see Figure 1D), an elegant study by Lessard et al. has characterized an essential change in subunit composition of a SWI/SNF-like chromatin remodeling complex during differentiation of NSCs to post-mitotic neurons (Lessard et al., 2007). Neural stem and progenitor cells express subunit proteins BAF45a and BAF53a as part of the SWI/SNF chromatin remodeling complex, which are replaced by BAF45b, BAF45c and BAF53b as progenitors exit the cell cycle. Importantly, the essential nature of this subunit change for neural differentiation was functionally validated. Taken together, the proteomic profiling and protein interactome studies of adult stem cells achieved thus far highlight the fact that these methodologies can and will lead to novel insights into the underlying cell biology that would not be discovered using other means.
3.2. Future directions of adult stem cell proteomics
The field of adult stem cell proteomics has a promising future. As new, improved and more sensitive methodologies become available, the limited numbers of obtainable adult stem cells will become less of a barrier. One very promising approach for a wide variety of applications in adult stem cell proteomic studies is use of protein microarrays or chips (see Figure 1E). These have the potential to identify protein-protein interactions, protein-phospholipid interactions, small molecule targets, and substrates of protein kinases, all while requiring a relatively small amount of starting material (Baharvand et al., 2007). The type of functional protein microarrays that have been used previously in yeast to study protein-protein interactions, specifically demonstrated to identify calmodulin binding proteins (Zhu et al., 2001), stand to be particularly valuable in characterizing the adult stem cell protein interactome.
One of the major issues facing adult stem cell proteomics, i.e., cell heterogeneity, ironically stands to be greatly aided by proteomic work itself. It has been demonstrated that isolation of what is considered to be an enriched hematopoietic stem/progenitor population (human umbilical cord blood CD34+ cells) still results in significant proteomic heterogeneity between samples (Zenzmaier et al., 2005). Adult stem cell populations expanded in vitro are also not immune to this issue, as it has been shown that human bone marrow MSC lines have divergent self-renewal and lineage differentiation capacities (Colter et al., 2001). The way in which proteomics will be able to address these issues is through further characterization of cell surface antigens expressed specifically on various adult stem cell types, which will allow even greater prospective isolation capability and thus more homogeneous cell populations. This has been recognized in the HSC field, where transcriptome profiling enabled identification of the SLAM family of cell surface markers (Kiel et al., 2005), which have improved means of HSC isolation. If transcriptome data is able to achieve moderate success to this end, there is vast potential to identify novel biomarkers through membrane proteomics. In addition, use of lineage-specific fluorescent reporters will allow isolation of more homogeneous cell populations. This strategy has been successfully employed in proteomic studies examining differentiation of mESCs to mesodermal/hemangioblast lineages with subsequent profiling using iTRAQ (Williamson et al., 2007).
4. Concluding remarks
The proteomics studies of embryonic, as well as adult, stem cells will complement characterization of these cells at the transcriptional level (transcriptome) and connect gene transcription and cellular phenotypes. The true challenge now is to integrate proteomics into the full spectrum of biological and biomedical research. Over the next decade, characterizing the proteome and interactome of stem cells through the identification of protein constituents, quantitation of protein concentration, dissection of protein interaction networks, and deciphering of transcriptional circuitry will provide a wealth of valuable information. These data will enable an integrated systems-level analysis and modeling of the mechanisms regulating stem cell self-renewal and potency. Combined advances in stem cell biology and MS hold great promise for dissecting components or pathways that either stimulate proliferation and self-renewal or induce differentiation towards specific cells or tissues. Ultimately, this will provide a framework for understanding the underlying biology of stem cells, and allow precise manipulation and realization of the full clinical therapeutic benefits of these unique cells.
Acknowledgements
This work is supported by Seed Grant from the Harvard Stem Cell Institute Cell Reprogramming Program to J.W., J.J.T. is a Leukemia & Lymphoma Society Fellow. S.R. is a NICHD Child Health Research Center Scholar and supported by a Career Development Award (K08) from the NHLBI. S.H.O. is an Investigator of Howard Hughes Medical Institute.