Hematopoiesis is sustained by a renewable pool of stem cells that interacts with distinct, sequential and specific microenvironments during normal development and throughout adult life. Hematopoietic stem cells (HSCs) are unique in their ability to migrate to various sites, ensuring the safety and integrity of their regenerative potential. This review is focused on the guidance cues and molecular pathways regulating HSC trafficking throughout the lifetime of the organism. We examine and discuss recent findings that shed new light into the molecular connections that feed a complex network between stem cells and their microenvironment, implicating parallel mechanisms for non-hematopoietic stem cells.
1. Introduction
Hematopoietic stem cell (HSC) migration throughout life is believed to be central to hematopoiesis under homeostasis. Blood circulation enables regulated trafficking of HSCs from specific embryonic and extra-embryonic sites to the fetal liver, ending their developmental journey in the bone marrow (BM) where most of the definitive lifelong hematopoiesis is maintained (Orkin and Zon, (2008)). However, HSCs continue to traffic throughout postnatal life for reasons that are not yet clear. Circulating HSCs can “home” to the bone marrow and lodge into specific microenvironments termed “niche” (French word for dog house), that allow their survival, self-renewal and regulated proliferation (Martinez-Agosto et al., (2007); Morrison and Spradling, (2008)). Conversely, BM HSCs egress constitutively into the bloodstream by a reverse phenomenon. Clinical HSC transplantation exploits this natural phenomenon through the enforced release of stem cells (referred to as “mobilization”) by cytokines, such as G-CSF, and/or chemotherapy to facilitate their collection in blood by leukapheresis. A simple intravenous infusion of HSCs/progeniors can reconstitute the BM hematopoietic reservoir after myeloablative therapy, significantly improving the clinical outcome of patients with a variety of diseases, especially in Oncology. Here, we review recent advances in our understanding of HSC trafficking during ontogeny and postnatal life. We suggest that mechanistic insight in HSC migration may serve as a template for other stem cells, particularly in cancer.
2. Trafficking of hematopoietic stem cells during embryogenesis and fetal development
2.1. HSC migration during the embryonic period
2.1.1. From the extraembryonic mesoderm to the embryo proper
The site of initial emergence of definitive HSCs among mammals has been controversial for over 30 years. Early erythroid, myeloid and lymphoid progenitor activity was found in the extraembryonic mesoderm of the yolk sac around embryonic day 7 (Moore and Metcalf, (1970); Palis et al., (1999); Samokhvalov et al., (2007)). However, other studies provide convincing evidence that definitive HSCs originate, by E8.5, from an intraembryonic site near the aorta, named the para-aortic splanchnopleura (P-Sp) which becomes the aorta-gonad-mesonephros (AGM) region (Godin and Cumano, (2002)). In addition, recent reports point to a third hematopoietic site, the allantoic mesoderm of the placenta, generating autonomously in situ a large pool of pluripotent HSCs in the mouse embryo between E8.5 and E9.5 (Alvarez-Silva et al., (2003); Gekas et al., (2005); Ottersbach and Dzierzak, (2005)) Although most studies have focused on the mouse system, the zebrafish model shares some notable similarities (Orkin and Zon, (2008)). For example, the site of initiation of definitive HSCs has been identified as a cluster of cells between the dorsal aorta and the posterior cardinal vein, anatomically homologous to the AGM region of mammals (Zhang and Rodaway, (2007)). The zebrafish AGM HSCs migrate to the caudal hematopoietic tissue (CHT) that mirrors the functions of both fetal liver and placenta in mammals, providing transient niche to support definitive HSC expansion and differentiation. Later these HSCs continue their migration toward the kidney serving, like the mammalian BM, as the definitive site of hematopoiesis for the adult life (Murayama et al., (2006)).
The development of a cardio-vascular network is probably as important for HSC trafficking as the invention of the wheel was for human travelling. In the mouse the first blood vessels are generated between embryonic days 6.5 to 9.5, a beating heart by E8.5, but a functional circulatory system is not achieved until E10, delaying the blood dispersal of HSCs into the embryo proper until E10.5 (Cumano et al., (2001); McGrath et al., (2003)). Recent studies in heartbeat deficient Ncx1−/- embryos, which do not survive beyond E10.5 due to the absence of functional circulation, suggest that HSCs may be independently generated in the placenta (Rhodes et al., (2008)). When the circulatory system becomes operative, definitive HSCs and myeloerythroid progenitors are capable to migrate from the embryonic hematopoietic sites through the circulation, starting their migratory journey by colonizing fetal liver at E10.5 (Mikkola and Orkin, (2006)).
2.1.2. Trafficking mechanisms in the embryo
Little is known, however, about the molecular mechanisms mediating HSC/progenitor trafficking during embryonic hematopoiesis. It appears that a microenvironment of mesenchymal origin already exists in the AGM, similar to that of the fetal liver or BM, since early mesodermal precursors have been isolated in the embryonic dorsal aorta (Mendes et al., (2005); Minasi et al., (2002)). The placental vessels may also provide a unique environment, as a vascular niche enriched in growth factors and cytokines, where HSCs may expand and differentiate prior to seeding the fetal liver (Rhodes et al., (2008)). Previous studies have found that the integrin α2b (GPIIb, CD41) marks HSCs in the embryo (Corbel and Salaun, (2002)). The α2bβ3 integrin heterodimer is a major receptor of fibrinogen on platelets. It role on HSCs is not clear since it appears to be dispensable for embryonic HSC adhesion, migration and proliferation; indeed, no overt developmental hematopoietic defects was noted in β3-deficient mice, which lack both α2bβ3 and αVβ3 (Hodivala-Dilke et al., (1999)). A possible role for VE-cadherin, an endothelial adhesion molecule, is also suggested by its expression on FLK1+ cells that contain definitive hematopoietic potential (Fraser et al., (2002)). VE-cadherin is downregulated when HSCs transit from embryonic sites to the fetal liver. Its downregulation does not appear to require contact with fetal liver cells, but may depend on the developmental stage (Taoudi et al., (2005)). The emergence of the circulation empowers HSCs to traffic among the embryonic sites and also colonize the fetal liver (see Figure 1). However, none of the knockout animals deficient in a single molecule mediating adhesion events of HSCs/progenitors under flow during fetal or adult life (e.g. selectins and α4 integrins, see below) has obvious deficits in fetal liver colonization, suggesting redundancy of adhesion mechanisms mediating embryonic HSC migration (Arroyo et al., (1999)). Consistent with this possibility, HSCs deficient in all β1 integrins exhibit profound, cell autonomous defects in the colonization of the liver and spleen but no hematopoietic defect in the yolk sac and AGM (Potocnik et al., (2000)). β1 integrin forms 12 distinct heterodimers with α1–11 and αV, but only three β1 integrins (α4β1, α5β1 and α6β1) are highly expressed on HSCs. The critical role of these integrins for cell migration on extracellular matrix, combined with the normal emergence of HSC activity in the yolk sac and AGM regions despite their absence, suggest that HSCs may be independently generated in these embryonic sites. This possibility is supported by recent studies in embryos that have no functional circulation (Rhodes et al., (2008)).
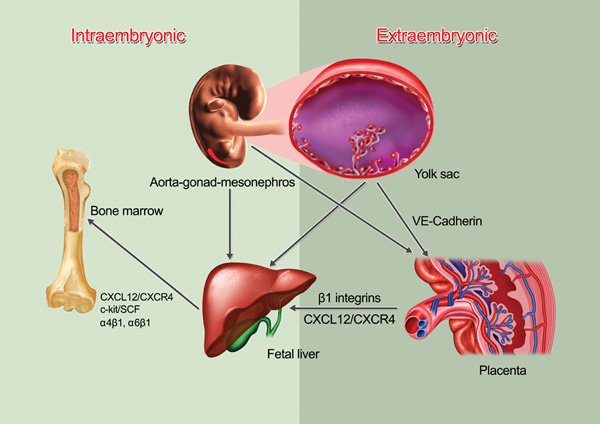
Figure 1. HSC migration from embryonic to fetal hematopoiesis. Definitive hematopoietic stem cell pools emerge from intra- (AGM) and extra- (yolk sac and placenta) embryonic hematopoietic sites. When the circulation is established and functional, HSCs start their migratory journey and colonize the placenta, followed by the fetal liver that becomes the major hematopoietic organ until the bone marrow colonized by HSCs. The adhesion molecule VE-cadherin expression has been suggested to participate in HSC/progenitor migration but there is no functional data about the molecular mediators of HSC migration in the embryo. The colonization of the fetal liver and the bone marrow are dependent on β1 integrins and the CXCL12/CXCR4 chemokine/receptor axis.
2.2. HSC migration during the fetal period
2.2.1. From the fetal liver to the bone marrow
In the course of ontogeny, the switch between sites of hematopoiesis is mediated by the migration of HSCs via the bloodstream. The liver, the primary fetal hematopoietic site, does not generate HSCs in situ but serves as a remarkable organ for expansion of HSCs that have migrated from embryonic sites (Johnson and Moore, (1975)). In parallel, during the third week of mouse gestation, mesenchymal progenitor cells that have the capacity to differentiate into cells of osteogenic, adipogenic and chondrogenic lineages, establish a unique microenvironment ideally suited for the colonization of the BM by HSCs from E17.5 (Mendes et al., (2005)). Shortly before birth, functional HSCs exit the fetal liver to migrate into the BM, when its environment is able to support their engraftment and self-renewal.
2.2.2. Trafficking mechanisms in the fetus
The microenvironmental cues involved in the shift of hematopoietic sites are not well understood. Among the possible regulatory factors derived from the microenvironment, stromal-derived factor-1 (SDF-1), more recently called CXCL12, and its cognate receptor CXCR4, have emerged as a master regulatory pathway of both HSC migration and retention in the bone marrow, during ontogeny and adult life (McGrath et al., (1999)). Indeed, myeloid progenitors are reduced in the BM and fetal liver of mice deficient in CXCR4 (Ma et al., (1998)). Similar defects were observed in the BM of CXCL12-deficient mice (Nagasawa et al., (1996)), suggesting that the CXCL12/CXCR4 axis is critical for fetal hematopoiesis. CXCL12 is expressed broadly during development and after birth, raising interesting questions about why it appears to be so specifically important for homing to fetal and adult hematopoietic sites. However, in situ hybridization studies of CXCL12 have revealed expression levels in the fetal liver and bone marrow that are concordant with the recruitment waves in these organs (McGrath et al., (1999)). In parallel, the CXCR4-dependent migratory capacity of progenitors also seems to follow a developmental switch (Aiuti et al., (1999)).
Fetal HSCs express high levels of c-kit on their surface and its ligand stem cell factor (SCF) is expressed in zones associated with migratory pathways of HSCs (Matsui et al., (1990)). SCF exerts both chemokinetic and chemotactic activites on HSCs/progenitors (Kim and Broxmeyer, (1998); Okumura et al., (1996)). In addition, it has been suggested that the chemotactic response to SCF is higher for fetal liver HSCs than adult BM HSCs, and that CXCL12 and SCF have synergistic effects on fetal liver HSC migration (Christensen et al., (2004)).
The integrins are the only family of adhesion molecules shown to be necessary for fetal hematopoiesis. As mentioned, α4 and β1 integrin expression is required for the colonization of the fetal liver and, consequently, of the fetal bone marrow. It is likely that the defect in the fetal BM originates from the impaired trafficking since conditional ablation of β1 integrins in adult HSCs abrogated the reconstitution of adult spleen and bone marrow (Potocnik et al., (2000)). Recent results implicate α6 integrin as a BM homing receptor for fetal liver progenitors, but not HSCs, whereas α4 integins displays a critical role in the migration of long-term repopulating HSCs in the adult BM (Qian et al., (2007)).
3. Post-natal HSC migration to bone marrow
The ability of HSC/progenitor to migrate continuously to the bone marrow was demonstrated decades ago in experiments in which shielding of the spleen allowed mice to recover from a lethal dose of irradiation (Jacobson et al., (1949)). These studies and others in the following decade (Barnes et al., (1956); Lorenz et al., (1951)) clearly demonstrated that splenic HSCs could spontaneously “home” to and repopulate the bone marrow, and paved the way toward the clinical use of bone marrow transplantation (Thomas, (1995)). Although the word “homing” has been used relatively loosely in the literature, it should refer to the initial interactions and migration of HSC/progenitors to the organ. “Lodgement” has often been used interchangeably with “homing”, but it implies a more definitive settling of stem cells in their niche. “Engraftment” refers to the ability of HSCs to proliferate, self-renew and give rise to multilineage progeny. HSC migration is a complex process involving a cascade of molecular events that are regulated centrally by the nervous system and enabled by the vasculature that permeates the bone marrow (see Plate 1). Each of the key steps has obvious consequences for successful clinical stem cell transplantation procedures. We will review below some of the mechanisms.
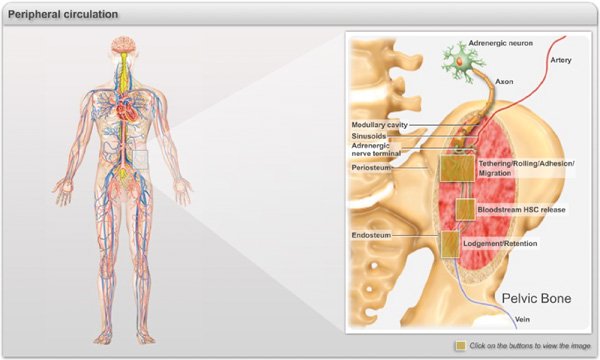
Plate 1. Main events of the journey of HSCs into the bone marrow during the adult life. Representative illustration of the events related to the hematopoietic trafficking occurring during the adulthood. HSCs circulate in the peripheral blood and enter into the bone marrow parenchyma through sinusoids and collecting venules via a cascade of molecular events commonly referred to as “homing” (Click on Box 1). After transmigration, HSCs/progenitors migrate through the extracellular matrix and the stromal elements to settle in specific niches near the vasculature and the endosteum (Click on Box 2). HSC/progenitors continuously egress into the blood stream in a circadian manner orchestrated by the sympathetic nervous system that regulates rhythmic fluctuation of CXCL12 expression in the bone marrow (Click on Box 3).
Box 1. Molecular basis of HSC/progenitor homing to bone marrow. A. HSCs are captured by and roll on E-selectin, P-selectins and Vascular cell adhesion molecule-1 (VCAM-1) which are constitutively expressed by endothelial cells of the bone marrow. P-selectin glycoprotein ligand-1 (PSGL-1) has been shown to be the major selectin ligand on HSCs but other ligands, such as CD44, contribute. HSC express high levels of α4β1 and also some α4β7, both which can mediate rolling on VCAM-1. B. Rolling interactions allow HSC to sample the chemokine CXCL12 presented in microdomains on the endothelial surface. CXCL12 can activate HSC through the G-protein coupled receptor CXCR4, leading to high affinity integrin-mediated arrest. C. Various adhesion molecules likely contribute to transendothelial and parenchymal migration, including α6β1 (laminin receptor), α5β1 (fibronectin receptor), CD99, CD31 and CD44 (as a hyaluronan receptor). CXCL12 may translocate from the endothelial cells to the parenchyma, affecting the migration of HSCs throughout BM. FLt3 may collaborate with CXCL12 to enhance migration of HSCs through the bone marrow matrix.
Box 2.Lodgement and retention of HSCs in the bone marrow niches (= box 3).Within the bone marrow, HSCs are attracted to CXCL12 rich regions near blood vessels and the endosteum where they lodge into niches that maintain their self-renewal and regenerative potential. To retain HSCs, the niches expression cell adhesion molecules (e.g. VCAM-1), matrix proteins (e.g. osteopontin) and grown factors (e.g. stem cell factor, angiopoietin-1), and chemokine (e.g. CXCL12) that regulate stem cell quiescence and attraction to the niche. The CXCL12/CXCR4 axis is under the control of a noradrenergic signals (NE) originating from sympathetic nerves modulating the synthesis of CXCL12 by stromal cells via the adrenergic receptor.
Box 3. Circadian release of HSCs in the peripheral circulation (= box 2). HSCs follow a physiologically regulated rhythmic release. Circadian HSC trafficking is orchestrated in central nervous system following inputs of light patterns that are processed in the suprachiasmatic nucleus (SCN), and entrained by the expression of core genes of the molecular clock. These signals are transmitted locally to bone marrow niches by rhythmic secretion of noradrenaline from nerve terminals that regulate HSC attraction through activation of the β3-adrenergic receptor (Adrβ3), degradation of Sp1, and downregulation of Cxcl12 transcription.
3.1. Initial interactions: homing
The extravasation of circulating HSCs within the BM requires a set of molecular interactions that mediates the recognition of circulating HSCs by the endothelium of BM sinusoids. Early work by Tavassoli and colleagues suggested that membrane lectin-carbohydrate interactions, notably involving galactosyl specificities, were involved between circulating HSCs and endothelium (Tavassoli and Hardy, (1990)). Integrins (e.g. α4β1) were then found to mediate the attachment of lymphoid progenitors to BM stromal cells (Miyake et al., (1991)). Papayannopoulou developed a seminal progenitor homing assay in adult mice and showed in 1995 that homing was significantly reduced (∼50%) by the inhibition of α4 integrins or VCAM-1 function (Papayannopoulou et al., (1995); see Box 1). The possible roles for lectins re-emerged when selectins were shown to be critical for the recruitment of mature leukocytes to inflamed sites (Frenette et al., (1996); Labow et al., (1994)). Indeed, in the absence of both endothelial selectins, E- and P-selectins, progenitor homing was compromised, particularly when VCAM-1 function was simultaneously inactivated (Frenette et al., (1998)). Intravital microscopy of adult bone marrow vasculature was developed at the same time, showing that α4β1/VCAM-1 and endothelial selectins contribute to the initial rolling interactions of progenitors on BM endothelium whereas α4β1/VCAM-1 is also involved in the arrest of progenitor cells (Mazo et al., (1998)). These studies revealed similarities between the multistep paradigm described for leukocytes and that of progenitors in that both roll on selectins prior to arrest on integrins and their immunoglobulin superfamily counter-receptors. However, further studies have revealed notable differences. For example, E-selectin closely collaborates with P-selectin in neutrophil recruitment to inflamed sites, whereas E-selectin cooperates with α4 integrins in the homing of long-term repopulating stem cells (Katayama et al., (2003)). The critical role of E-selectin ligands and α4 integrins in HSC homing is consistent with the constitutive expression of their counter-receptors, VCAM-1 and E-selectin, on bone marrow endothelium (Schweitzer et al., (1996)). Interestingly, recent studies using advanced imaging technologies have revealed that E-selectin colocalized with CXCL12 into specific microdomains in bone marrow venules where cancerous and HSC/progenitor cells extravasate (Sipkins et al., (2005)).
Analyses of selectin ligand expression and function have uncovered developmental regulation of key glycosylation enzymes that synthesize selectin ligands. For example, analyses of human CD34+ cell behavior by intravital microscopy have revealed reduced selectin mediated rolling of neonate (cord blood)-derived CD34+ progenitors compared to adult CD34+ cells obtained from the BM or peripheral blood after G-CSF stimulation (Hidalgo et al., (2002)). This deficiency is due to reduced fucosyltransferase activity in neonatal CD34+ cells (Hidalgo and Frenette, (2005); Xia et al., (2004)). Although enforced fucosylation can clearly enhance progenitor homing, whether this translates in improved engraftment is controversial, with one study providing positive results (Xia et al., (2004)) and one negative study (Hidalgo and Frenette, (2005)). The difference may be due to the variability of NOD/SCID mouse engraftment by human SCID-repopulating cells, or alternatively to the possible toxicity of Mn2+ exposure during the fucosylation protocol (Sackstein et al., (2008)). Enforced fucosylation using a Mn2+-free protocol endowed mesenchymal progenior cells with BM homing ability by converting CD44 into a bona fide E-selectin ligand (Sackstein et al., (2008)). CD44 is widely expressed in various tissues but is unable to bind to E-selectin unless properly fucosylated. CD44 expressed on HSC/progenitor cells (Dimitroff et al., (2001)) and mature myeloid cells (Katayama et al., (2005)), is able to bind to E-selectin. CD44 may also contribute to progenitor homing by interacting with hyaluronic acid (Avigdor et al., (2004); Vermeulen et al., (1998)). In addition to collaborating with E-selectin, α4 integrins synergize with β2 integrins during the initial endothelial capture of HSCs to the BM (Papayannopoulou et al., (2001)). Further studies into the receptors have revealed that both α4 integrins, α4β1 and α4β7, are expressed on HSCs and contribute equally to progenitor homing (Katayama et al., (2004)). A recent study also suggests that α4 integrins can act synergistically in vivo with α6 integrins as HSCs homing receptors (Qian et al., (2006)). Since α6 integrins are laminin receptors, it is likely that they participate at later steps of the homing cascade, during or after transmigration (see Box 1).
The coordinated action of these adhesion molecules is triggered by the chemokine CXCL12 presented at the surface of endothelial cells. Blockade of its receptor, CXCR4, was shown to inhibit human CD34+CD38- immature human progenitor homing and engraftment in NOD/SCID mice (Peled et al., (1999)). CXCL12 mediates activation of αLβ2 (Lymphocyte function-associated antigen-1, LFA-1 or CD11a/CD18), α4 and α5β1 (VLA-5 or CD49e/CD29) integrins, converting the rolling of CD34+ cells into stable arrest on the endothelium (Peled et al., (2000)). Downstream signals from CXCR4 involve the activation of PI-PLC and PI3K, that activate PKC-ζ which in turn, will induce Pyk2 and ERK activation, leading to adhesion and chemotaxis of progenitors (Bonig et al., (2004); Petit et al., (2005)). In addition, the CXCL12/CXCR4 axis crosstalks with other pathways that may enhance the migration of HSCs. For example, the combination of Flt3 and CXCL12 acts synergistically in the migration of CD34+ cells. In contrast, prolonged exposure to Flt3 may down-regulate CXCR4 expression and impair the migration of CD34+ cells toward CXCL12 (Fukuda et al., (2005)). Following firm adhesion, CXCL12 induces an integrin-dependent transmigration of progenitors across the endothelial lining, a phenomenon referred to as diapedesis. Adhesion molecules, such as platelet-endothelial cell adhesion molecule-1 (CD31/PCAM-1; Yong et al., (1998)) and CD99 (Imbert et al., (2006)), expressed on HSCs, may be involved in diapedesis in response to CXCL12. Although the site of transmigration has never been documented, it is possible that HSCs may use endothelial fenestration to enter the bone marrow parenchyma. Such transcellular migration has been described for the egress of mature blood cells from the marrow (Chamberlain and Lichtman, (1978)).
3.2. Mechanisms of lodgement and retention in the stem cell niche
3.2.1. Lodgement in bone marrow niches
Once crossed the endothelial sinusoids, HSCs migrate through the ECM within the BM parenchyma to lodge in a suitable location for survival, proliferation and differentiation. Given the inherent difficulties in tracking HSC/progenitor in the BM microenvironment, little is known about the mechanism of migration in the parenchyma. One study addressed this issue by evaluating the differential engraftment of human CD34+CD38- stem cells injected either directly in the marrow or intravenously, and found that the CXCR4 receptor was necessary for SCID-repopulating cell engraftment even when the homing step was bypassed by intra-marrow injection (Yahata et al., (2003)). The precise spatial localization of HSCs is controversial (Kiel and Morrison, (2008); also see Box 2). Serial sections of fluorescently labelled lineage negative cells have indicated that progenitors tend to localize near the endosteum (Nilsson et al., (2001)). This observation was consistent with earlier studies suggesting that CFU-S lodged predominantly in the endosteal region of femoral bones (Gong, (1978); Lord et al., (1975)). More recent studies using BrdU labelling have suggested that label-retaining slow-cycling stem cells were localized near spindle-shaped N-cadherin expressing osteoblasts, and that an increase in the osteoblast (OB) activity, such as the conditional inactivation of BmprIa or the administration of parathyroid hormone (PTH), could increase the number of HSCs in bone marrow (Calvi et al., (2003); Zhang et al., (2003)). PTH has been reported to increase the expression of CXCL12 by osteoblasts, which may retain HSCs in their endosteal niches (Jung et al., (2006)). HSC may sense extracellular calcium-ion concentration through the calcium receptor (CaR), whose expression is important for localization in the endosteum (Adams et al., (2006)). Other studies suggest that the transmembrane form of stem cell factor (tm-SCF) plays an important role in the lodgement into the endosteal niche (Driessen et al., (2003)). A role for OB activity in the regulation of HSC behavior is further supported by the fact that cultured OBs can expand the number of HSCs in vitro and in vivo (El-Badri et al., (1998); Taichman et al., (1996)), and by the specific ablation of OBs through a suicide gene strategy which dramatically affects BM hematopoiesis (Visnjic et al., (2004)). OBs secrete angiopoietin-1 which can induce HSC quiescence through its receptor Tie2 (Arai et al., (2004)), thus providing a molecular explanation for the relative quiescence in the OB niche. However, BrdU retention as a stem cell marker and the role of N-cadherin are controversial (Kiel et al., (2007); Kiel et al., (2007)). Recent follow-up studies suggest that the levels of N-cadherin may distinguish two HSC populations that are primed to migrate out of the niche (low expression levels) or be kept in “reserve” (intermediate levels; Haug et al., (2008)). The role of OBs has been challenged with the analyses of mice lacking biglycan which are reported to have fewer OBs, but exhibit no changes in the number of stem cells (Kiel et al., 2007). It is possible that the difference between these studies may originate from the models and the stage of OB differentiation since HSCs are extremely rare and therefore must be associated with a small subset of OBs.
The identification of novel stem cell markers by SLAM expression (CD150+CD48- cells) has revealed that ∼70% of CD150+CD48- HSCs were found near blood vessels (Kiel et al., (2005); also see Box 2). Like OBs, previous studies have indicated that cultured endothelial cells support hematopoiesis and increase the repopulating ability of BM CD34+ cells in NOD/SCID mice (Chute et al., (2002); Davis et al., (1995); Li et al., (2004)). Interaction with sinusoidal endothelium at the vascular niche has been suggested to induce hematopoietic cell expansion, differentiation in vitro and in vivo and egress of mature progenitors (Kopp et al., (2005)). For example, the growth factor thrombopoietin (TPO) is known to stimulate the early development of hematopoietic lineage and megakaryopoiesis (Kaushansky, (1995)). Targeting this gene or the gene coding for its receptor (c-mpl) induces thrombocytopenia (Gurney et al., (1994)). CXCL12 and FGF-4 can restore thrombopoiesis in TPO- or c-mpl-deficient mice by stimulation of VE-Cadherin-, VLA-4/VCAM-1-mediated localization of CXCR4+ megakaryocyte progenitors to the vascular niche (Avecilla et al., (2004)). Additional studies have demonstrated that the tyrosine kinase Tie2 receptor expressed on endothelium and on hematopoietic cells is implicated in the assembly and remodelling of BM neovessels after myelosuppression. Inhibition of Tie2 impairs neoangiogenesis, leading to a delay in hematopoietic recovery. Conversely, Angiopoietin-1 stimulates hematopoiesis in TPO-deficient mice (Kopp et al., (2005)). The vascular niche could thus be viewed as a dynamic scaffold that allows rapid hematopoietic cell production. Active proliferation of progenitors around the vascular niche has led to the suggestion that HSCs localized near blood vessels might be proliferative whereas HSC in the endosteum are quiescent. Although progenitors are clearly proliferating near the vasculature, there is currently no experimental evidence supporting the notion that vessel-associated HSCs are less quiescent than those located near the endosteum.
3.2.2. Factors contributing to HSC retention and quiescence
Following the lodgement into a suitable niche, HSCs may undergo a series of symmetrical division to expand their numbers, but then HSC must transition from active cycling to quiescence while maintaining a delicate balance between self-renewal and lineage commitment. The matrix product of osteoblasts osteopontin (Opn), which can interact with CD44 and β1 integrins on HSCs, has been suggested to regulate negatively the proliferation and differentiation of HSCs in the niche. Opn−/- mice exhibit markedly enhanced cycling of HSCs (Nilsson et al., (2005); Stier et al., (2005)). The receptor tyrosine kinase Tie2 expressed on HSCs interacts with its ligand angiopoietin-1 expressed by OBs. This interaction maintains long-term repopulating activity of HSCs in vivo, inducing HSCs quiescence and adhesion to the niche (Arai et al., (2004)). Similarly, adhesive contacts through N-cadherin or c-kit/SCF interactions may also hold HSCs anchored to the endosteal niche, promoting their quiescence (Arai et al., (2004); Driessen et al., (2003); Haylock and Nilsson, (2005)). A recent study suggests that CXCR4/CXCL12 signaling is critical for HSC quiescence under homeostasis (Nie et al., (2008)). Upon ablation of c-Myc, a cell intrinsic regulator of quiescence, HSCs remained quiescent, undifferentiated and anchored to the niche as adhesion molecule expression increased on their surface. Conversely, enforced c-Myc expression in HSCs downregulates N-cadherin and integrins, leading of a loss of self-renewal at the expense of differentiation (Wilson et al., (2004)).
The role for α4 integrins (α4 is encoded by Itga4) and their ligand VCAM-1 in HSC retention was evaluated with mice conditionally deficient in either Itga4 or Vcam1. Itga4-deficiency induced by Mx.cre deletion rapidly increased the number of circulating progenitors in blood suggesting a role in BM retention (Scott et al., (2003)). Similar results were noted in mice in which Vcam1 was deleted by Tie2-driven cre transgene (Ulyanova et al., (2005)). Downstream signals from the Rho GTPases Rac1 and Rac2, regulating actin assembly and motility, are critical in HSC retention as their levels are increased in the circulation in deficient animals (Cancelas et al., (2005)). In these models, however, it is difficult to distinguish the contributions of each molecule in the retention versus homing to the bone marrow since HSCs continually recirculate in blood (see below). Other integrins may also participate in retention. For example, αMβ2 which is expressed on progenitors and activated HSCs (Morrison et al., (1997)), appears to retain progenitors in situations where the egress is enforced (Hidalgo et al., (2004)).
Among the factors that contribute to HSC retention in their niches, CXCL12 has emerged has a critical regulator. Although previous studies have indicated that OBs and endothelial cells synthesize CXCL12, studies using Cxcl12 gene knockin have indicated that the stromal cells expressing the highest levels of CXCL12 are reticular cells (Tokoyoda et al., (2004)). These CXCL12-abundant reticular (CAR) cells are scattered throughout the bone marrow and surrounds sinusoidal endothelial cells (Sugiyama et al., (2006)). Recently, CD146+ human stromal progenitors were identified and defined as generating adventitial reticular cells and osteoblast progenitors and then are capable to establish a unique hematopoietic microenvironment in vivo (Sacchetti et al., (2007)). In the absence of the CXCL12 receptor CXCR4, progenitors are released prematurely in the blood of adult mice (Foudi et al., (2006); Ma et al., (1999)). Desensitization of the receptor using a N-terminal methionine analog of CXCL12 increased the number of circulating Lineage- Thy-1low Sca-1+ c-kit+ cells in blood (Shen et al., (2001)). Under hypoxic microenvironment, CXCL12 can be regulated by hypoxia inducible factor-1 (HIF-1; Ceradini et al., (2004)). HSC have been suggested to localize in relatively hypoxic areas (Parmar et al., (2007)). However, the location of CAR cells near blood vessels (an area presumably adequately oxygenated), suggests that HIF-1 may play a more prominent role in pathological situation, and that other transcription factors may regulate physiological CXCL12 expression. Notably, the Cxcl12 promoter has several highly conserved Sp binding sites (Garcia-Moruja et al., (2005)). Recent studies indeed suggest that circadian degradation of the transcription factor Sp1 is under the control of β3-adrenergic signalling which regulates CXCL12 expression (Mendez-Ferrer et al., (2008)). A reduction of CXCL12 expression within the BM microenvironment has been associated with virtually all methods of HSC mobilization that are discussed in the next section.
4. Egress of bone marrow HSCs to the periphery
The constitutive presence of HSCs in the circulation, or egress from sites of hematopoiesis, was inferred several decades ago through studies of parabiotic mice (partners that share the blood circulation) in which the lethally irradiated partner could survive a lethal dose of irradiation (Brecher and Cronkite, (1951); Warren et al., (1960)). The detection of progenitor cell activity was further substantiated with the development of assays in secondary recipient or directly in culture (Goodman and Hodgson, (1962); McCredie et al., (1971)). It was soon noted that circulating progenitors were significantly mobilized into the bloodstream from chemotherapy (Richman et al., (1976)). This observation, combined with the development of leukopheresis techniques, propagated the use of peripheral blood stem cells for transplantation. During the analyses of recombinant hematopoietic cytokines to support hematopoiesis during cancer therapy, it was noted that granulocyte colony-stimulating factor (G-CSF) increased significantly the number of progenitors in the circulation (Duhrsen et al., (1988)). The potential of G-CSF to elicit circulating stem cells for transplantation was demonstrated in the early 1990's (Molineux et al., (1990)), and soon adopted as the most powerful and safe stem cell mobilizer in the clinic. However, in a significant subset of patients, particularly those that have previously received cytotoxic or radiation therapy, HSCs recovery from the blood may not be sufficient for autologous transplantation. This underscores the importance of understanding the natural mechanisms of HSC egress and the pathways implicated in the therapeutic mobilization of HSCs to the circulation.
4.1. Physiological HSC egress into the bloodstream
The presence of circulating HSCs in blood under homeostasis suggests a turnover in BM niches. Decades old parabiotic techniques were elegantly resurrected with modern HSC markers to assess this issue (Wright et al., (2001)). These studies revealed that following 7 weeks of parabiosis, ∼5–10% of HSCs are derived from the partner. That and the fact that isolated HSCs injected in wild-type recipients are cleared very rapidly from the circulation, suggested a high steady-state HSC trafficking in blood. However, it is possible that the ex vivo manipulation of HSCs may have accelerated their clearance and potentially overestimated the actual turnover. HSC release in the circulation follow a circadian pattern where blood HSCs reach a peak in mice 5h after the onset of light and then reach a nadir 5h after darkness (Mendez-Ferrer et al., (2008)). Remarkably, blood HSCs fluctuate in antiphase with CXCL12 expression in the bone marrow environment, which is regulated at the mRNA level by rhythmic release of noradrenaline by nerve terminals of the sympathetic nervous system. Under physiological conditions, adrenergic signals, transduced specifically by the β3 adrenoreceptor, play a major role in controlling Cxcl12 expression in the bone marrow microenvironment by regulating the degradation of the transcription factor Sp1 in the nucleus of stromal cells (see Box 3). Although the role of circulating HSC in physiology is unclear at the moment, the peak release during the murine resting period argues for a role in the regeneration of the niche. HSC have been recovered in peripheral tissues like muscles (McKinney-Freeman et al., (2002)). HSCs also traffic in extramedullary sites such as the lymph, suggesting that they might contribute to immune cells in situ (Massberg et al., (2007)). Further studies are needed to determine whether the natural rhythm of HSCs release might be exploited in humans to optimize the yield and facilitate HSC collection for transplantation.
4.2. Mechanisms of G-CSF-induced mobilization
G-CSF, the most commonly used agent in the clinical arena, can elicit robust mobilization in 5–10 days. Owing to its widespread use, mobilization by G-CSF has served as the prototype to obtain mechanistic insights into this phenomenon. Predictably, mice deficient in the G-CSF receptor (encoded by Csf3r) are unresponsive to G-CSF treatment; Csf3r−/- HSCs can be mobilized by G-CSF in chimeric mice harboring mixtures of Csf3r+/+ and Csf3r−/- hematopoietic cells. This suggests that expression of the CSF3R on HSCs is not required for G-CSF-mediated mobilization and support a model in which CSF3R-dependent signals act in trans (Liu et al., (2000)). Subsequent studies have suggested that metallo- and/or serine proteases may represent the soluble “signal” released in G-CSF mobilization. Indeed, G-CSF induces the activation of various proteases, most notably neutrophil elastase and cathepsin G, which were shown to cleave VCAM-1 in the BM microenvironment (Levesque et al., (2001)). Further studies revealed that CXCL12 and CXCR4 were also targeted by BM proteolytic activity (Levesque et al., (2003); Petit et al., (2002); Semerad et al., (2002)), and an elastase inhibitor blocked G-CSF mobilization (Petit et al., (2002)). The release of membrane bound SCF through metalloproteinase-9 (MMP-9) activity was proposed to contribute to G-CSF mobilization since it was inhibited in Mmp9−/- mice (Heissig et al., (2002)). Others studies, however, have found no defect in G-CSF-induced mobilization in Mmp9−/- mice (Papayannopoulou et al., (2003); Pelus et al., (2004)). Another class of protease, dipeptidylpeptidase IV (CD26), expressed on a subset of HSCs and capable of cleaving the functional N-terminal segment of CXCL12, was also proposed to mediate CXCL12 inactivation and mobilization (Christopherson et al., (2003)). In parallel, concentrations of Serpin1 and Serpin3 in the BM dramatically drop following G-CSF administration, shifting the balance between proteases and their inhibitors (Winkler et al., (2005)). However, G-CSF-induced mobilization was normal in mice lacking virtually all neutrophil serine protease activity (dipeptidylpeptidase-I-deficient), even when combined with a broad metalloproteinase inhibitor, suggesting that other mechanisms must be involved (Levesque et al., (2004)).
Recent studies using various pharmacologic and genetic models have revealed that signals emanating from the sympathetic nervous system are critical for G-CSF-induced mobilization (Katayama et al., (2006)). Indeed, CXCL12 expression was dysregulated following G-CSF administration in mice that have severe abnormalities in nerve conduction (ceramide galactosyltransferase-deficient or Cgt−/-). CXCL12 is stored in the bone matrix and its rapid downregulation by both translational and posttranslational mechanisms is critical for optimal egress of HSCs from the BM. Bone CXCL12 is not downregulated in Cgt−/- mice following G-CSF administration suggesting that it may act like a magnet retaining HSCs and progenitors in the BM microenvironment. The sympathetic branch of the nervous system plays an important role because its disruption significantly impaired the effect of G-CSF. Although the administration of a β2 adrenergic agonist enhances mobilization induced by G-CSF, it is not sufficient to mobilize by itself, suggesting the contribution of other signals. As mentioned, adrenergic signalling through the β3 receptor is sufficient to regulate Cxcl12 mRNA expression under physiological steady-state (Mendez-Ferrer et al., (2008)). The involvement of the sympathetic nervous system in HSC mobilization is consistent with an emerging theme where stress signals can lead to HSC egress. G-CSF may modulate neural activity directly, as suggested by the expression of CSF3R on neuron and the effect of G-CSF on neuronal survival (Schneider et al., (2005)) and protects dopaminergic neurons in a model of Parkinson's disease (Meuer et al., (2006)). However, studies in transplantation chimeras suggest that CSF3R expression on a transplantable hematopoietic cell is required for efficient G-CSF-induced mobilization (Liu et al., (2000)). Components of innate immunity may participate in HSC/progenitor retention since mice deficient in C3 or C3a receptor are more sensitive to G-CSF mobilization (Ratajczak et al., (2004)). The mechanisms by which G-CSF induces HSC trafficking are thus complex and may involve more than one cellular targets, which have not been clearly defined.
4.3. Mobilization induced by other molecules
Several other compounds or drugs have been shown to trigger HSC egress from the marrow. When known, the mechanisms point, either directly or indirectly, toward the disruption of CXCL12−CXCR4 function. Cytotoxic drugs like cyclophosphamide, the first class of molecules found to mobilize HSCs, also require intact CSF3R expression (Liu et al., (1997)) and lead to increased proteolytic activity and reduced CXCL12 levels in the BM microenvironment (Levesque et al., (2002)). Several cytokines have been suggested to increase the number of circulating progenitors in blood, including IL-1, IL-3, IL-6, IL-7, IL-8, IL-11, IL-12, GM-CSF, SCF, Mip-1α, and Groβ (reviewed in Cottler-Fox et al., (2003)). Counter-intuitively, both an agonist (Pelus et al., (2005)) and an antagonist of CXCR4 (AMD3100) can enhance mobilization when used in combination with G-CSF (Broxmeyer et al., (2005); Devine et al., (2004); Flomenberg et al., (2005); Liles et al., (2003)). Increased plasma levels of CXCL12 can also mobilize human progenitors in NOD/SCID mice (Perez et al., (2004)). Administration of negatively charged polymers, such as fucoidan, leads to rapid progenitor mobilization (Frenette and Weiss, (2000)), likely through changes in membrane-bound CXCL12 (Sweeney et al., (2002)). Anti-adhesion therapy against α4 integrins was recently shown to enhance circulating progenitors in a clinical trial (Bonig et al., (2008)). Based on pre-clinical studies, this effect would likely be enhanced by blocking β2 integrins or both endothelial selectins (Katayama et al., (2003); Papayannopoulou et al., (2001)). Thus blocking of homing pathways together with factors that promote egress would represent a powerful strategy to enhance mobilization.
Another strategy would be to modulate the number of functional HSC niches. For example, parathyroid hormone administration for 5 weeks to stimulate bone formation and increase the number of HSC niches, enhanced the number of G-CSF-mobilized HSCs/progenitors (Adams et al., (2007)). More acutely, osteoclast activation resulting from a stress (e.g. severe bleed) has been suggested to mobilize progenitors (Kollet et al., (2006)). Administration of the biphosphonate pamidronate, which blocks osteoclast-mediated bone destruction, unexpectedly enhances G-CSF-induced HSC mobilization (Takamatsu et al., (1998)). It will be interesting to evaluate the role of β2/β3 agonists in clinical trials in patients predicted to mobilize HSCs poorly (Katayama et al., (2006); Mendez-Ferrer et al., (2008)).
5. Perspectives
HSCs integrate many signals that determine their trafficking, localization and subsequently their function. Under homeostasis, these signals involve the regulation of adhesive interactions, expression of matrix-degrading enzymes, cytoskeletal rearrangements, chemokinesis and chemotaxis, which are tightly regulated. It is not clear whether, and if so, how these different activities are interconnected to maintain a calibrated HSC movement. Recent studies suggest that these signals may work together in a complex network within which the CXCL12/CXCR4 chemotactic axis appears to play a prominent role. Further studies are needed to understand better the differential functions, if any, of the vascular and osteoblastic niches. In addition, little is known about the trafficking inside the bone marrow microenvironment, toward putative lineage-specific or possibly other stem cell niches. Evidence for specialized sous-niches already exits. For example, OB may also play an important role in lymphoid commitment (Katayama et al., (2006); Zhu et al., (2007)) whereas the macrophage brings a nurturing environment forming erythroblast islands that provide a scaffold for enucleation (Bessis, (1958); Soni et al., (2006)), and megakaryocyte differentiation takes place prominently near blood vessels (Kopp et al., 2005 ).
Considering the potential parallel between normal and cancer stem cells, it is possible that leukemic stem cells (LSC) settle in HSC niches, and usurp similar trafficking machinery (Ninomiya et al., (2007); Sipkins et al., (2005)). G-CSF-mobilized purified HSC fractions are contaminated by leukemic cells, suggesting that they can egress via the same stimuli (Isidori et al., (2007)). Mice deficient in the tumor suppressor Pten show an impaired lodgement and increased mobilization of HSCs and proliferation of leukemic stem cells, confirming a close relationship between intrinsic survival pathways and extrinsic cues from the niche (Zhang et al., (2006)). A similar parallel may be operative for stem cells of solid tumors, as suggested by the fact that breast carcinoma and prostate cancer, which preferentially metastasize to the bone, use CXCR4/CXCL12 to migrate (Balkwill, (2004)). The field of cancer biology may indeed benefit greatly from general advances in stem cell trafficking.
Acknowledgments
We thank the members of the Frenette laboratory (Drs. Andres Hidalgo, Daniel Lucas and Simon Mendes-Ferrer) for helpful comments on the manuscript. C.M. was supported in part by a fellowship from the French Fondation pour la Recherche Médicale. P.S.F. is supported by the National Institutes of Health, the Department of Defense, and the American Heart Association.
Copyright: © 2008 Claire Magnon and Paul S. Frenette.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
§ To whom correspondence should be addressed. E-mail: paul.frenette@mssm.edu
* Edited by: David Scadden. Last revised April 29, 2008. Published July 14, 2008. This chapter should be cited as: Magnon, C. and Frenette, P.S., Hematopoietic stem cell trafficking (July 14, 2008), StemBook, ed. The Stem Cell Research Community, StemBook, doi/10.3824/stembook.1.8.1, https://www.stembook.org .
References
- Adams, G. B. Chabner, K. T. Alley, I. R. Olson, D. P. Szczepiorkowski, Z. M. Poznansky, M. C. Kos, C. H. Pollak, M. R. Brown, E. M. Scadden, D. T. (2006). Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 439, 599–603. Abstract DOI
- Adams, G. B. Martin, R. P. Alley, I. R. Chabner, K. T. Cohen, K. S. Calvi, L. M. Kronenberg, H. M. Scadden, D. T. (2007). Therapeutic targeting of a stem cell niche. Nat Biotechnol 25, 238–243. Abstract DOI
- Aiuti, A. Tavian, M. Cipponi, A. Ficara, F. Zappone, E. Hoxie, J. Peault, B. Bordignon, C. (1999). Expression of CXCR4, the receptor for stromal cell-derived factor-1 on fetal and adult human lympho-hematopoietic progenitors. Eur J Immunol 29, 1823–1831. Abstract DOI
- Alvarez-Silva, M. Belo-Diabangouaya, P. Salaun, J. Dieterlen-Lievre, F. (2003). Mouse placenta is a major hematopoietic organ. Development 130, 5437–5444. Abstract DOI
- Arai, F. Hirao, A. Ohmura, M. Sato, H. Matsuoka, S. Takubo, K. Ito, K. Koh, G. Y. Suda, T. (2004). Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118, 149–161. Abstract DOI
- Arroyo, A. G. Yang, J. T. Rayburn, H. Hynes, R. O. (1999). Alpha4 integrins regulate the proliferation/differentiation balance of multilineage hematopoietic progenitors in vivo. Immunity 11, 555–566. Abstract DOI
- Avecilla, S. T. Hattori, K. Heissig, B. Tejada, R. Liao, F. Shido, K. Jin, D. K. Dias, S. Zhang, F. Hartman, T. E. (2004). Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med 10, 64–71. Abstract DOI
- Avigdor, A. Goichberg, P. Shivtiel, S. Dar, A. Peled, A. Samira, S. Kollet, O. Hershkoviz, R. Alon, R. Hardan, I. (2004). CD44 and hyaluronic acid cooperate with SDF-1 in the trafficking of human CD34+ stem/progenitor cells to bone marrow. Blood 103, 2981–2989. Abstract DOI
- Balkwill, F. (2004). Cancer and the chemokine network. Nat Rev Cancer 4, 540–550. Abstract DOI
- Barnes, D. W. H. Corp, M. J. Loutit, J. F. Neal, F. E. (1956). Treatment of murine leukaemia with x-rays and homologous bone marrow. Preliminary communication. Br Med J 2, 626–627.
- Bessis, M. (1958). [Erythroblastic island, functional unity of bone marrow.]. ] Rev Hematol 13, 8–11. Abstract Abstract
- Bonig, H. Priestley, G. V. Nilsson, L. M. Jiang, Y. Papayannopoulou, T. (2004). PTX-sensitive signals in bone marrow homing of fetal and adult hematopoietic progenitor cells. Blood 104, 2299–2306. Abstract DOI
- Bonig, H. Wundes, A. Chang, K. H. Lucas, S. Papayannopoulou, T. (2008). Increased numbers of circulating hematopoietic stem/progenitor cells are chronically maintained in patients treated with the CD49 d blocking antibody natalizumab. Blood 111, 3439–3441. Abstract DOI
- Brecher, G. Cronkite, E. P. (1951). Post-radiation parabiosis and survival in rats. Proc Soc Exp Biol Med 77, 292–294. Abstract Abstract
- Broxmeyer, H. E. Orschell, C. M. Clapp, D. W. Hangoc, G. Cooper, S. Plett, P. A. Liles, W. C. Li, X. Graham-Evans, B. Campbell, T. B. (2005). Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med 201, 1307–1318. Abstract DOI
- Calvi, L. M. Adams, G. B. Weibrecht, K. W. Weber, J. M. Olson, D. P. Knight, M. C. Martin, R. P. Schipani, E. Divieti, P. Bringhurst, F. R. (2003). Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846. Abstract DOI
- Cancelas, J. A. Lee, A. W. Prabhakar, R. Stringer, K. F. Zheng, Y. Williams, D. A. (2005). Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat Med 11, 886–891. Abstract DOI
- Ceradini, D. J. Kulkarni, A. R. Callaghan, M. J. Tepper, O. M. Bastidas, N. Kleinman, M. E. Capla, J. M. Galiano, R. D. Levine, J. P. Gurtner, G. C. (2004). Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10, 858–864. Abstract DOI
- Chamberlain, J. K. Lichtman, M. A. (1978). Marrow cell egress: specificity of the site of penetration into the sinus. Blood 52, 959–968.
- Christensen, J. L. Wright, D. E. Wagers, A. J. Weissman, I. L. (2004). Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol 2, E75. Abstract DOI
- Christopherson, K. W. 2nd, Cooper, S. Broxmeyer, H. E. (2003). Cell surface peptidase CD26/DPPIV mediates G-CSF mobilization of mouse progenitor cells. Blood 101, 4680–4686. Abstract DOI
- Chute, J. P. Saini, A. A. Chute, D. J. Wells, M. R. Clark, W. B. Harlan, D. M. Park, J. Stull, M. K. Civin, C. Davis, T. A. (2002). Ex vivo culture with human brain endothelial cells increases the SCID-repopulating capacity of adult human bone marrow. Blood 100, 4433–4439. Abstract DOI
- Corbel, C. Salaun, J. (2002). AlphaIIb integrin expression during development of the murine hemopoietic system. Dev Biol 243, 301–311. Abstract DOI
- Cottler-Fox, M. H. Lapidot, T. Petit, I. Kollet, O. DiPersio, J. F. Link, D. Devine, S. (2003). Stem cell mobilization. Hematology Am Soc Hematol Educ Program , 419–437. Abstract Abstract
- Cumano, A. Ferraz, J. C. Klaine, M. Di Santo, J. P. Godin, I. (2001). Intraembryonic, but not yolk sac hematopoietic precursors, isolated before circulation, provide long-term multilineage reconstitution. Immunity 15, 477–485. Abstract DOI
- Davis, T. A. Robinson, D. H. Lee, K. P. Kessler, S. W. (1995). Porcine brain microvascular endothelial cells support the in vitro expansion of human primitive hematopoietic bone marrow progenitor cells with a high replating potential: requirement for cell-to-cell interactions and colony-stimulating factors. Blood 85, 1751–1761. Abstract Abstract
- Devine, S. M. Flomenberg, N. Vesole, D. H. Liesveld, J. Weisdorf, D. Badel, K. Calandra, G. DiPersio, J. F. (2004). Rapid mobilization of CD34+ cells following administration of the CXCR4 antagonist AMD3100 to patients with multiple myeloma and non-Hodgkin's lymphoma. Clin Oncol 22, 1095–1102. Abstract DOI
- Dimitroff, C. J. Lee, J. Y. Rafii, S. Fuhlbrigge, R. C. Sackstein, R. (2001). CD44 is a major E-selectin ligand on human hematopoietic progenitor cells. J Cell Biol 153, 1277–1286. Abstract DOI
- Driessen, R. L. Johnston, H. M. Nilsson, S. K. (2003). Membrane-bound stem cell factor is a key regulator in the initial lodgment of stem cells within the endosteal marrow region. Exp Hematol 31, 1284–1291. Abstract DOI
- Duhrsen, U. Villeval, J. L. Boyd, J. Kannourakis, G. Morstyn, G. Metcalf, D. (1988). Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood 72, 2074–2081. Abstract Abstract
- El-Badri, N. S. Wang, B. Y., Cherry. Good, R. A. (1998). Osteoblasts promote engraftment of allogeneic hematopoietic stem cells. Exp Hematol 26, 110–116. Abstract Abstract
- Flomenberg, N. Devine, S. M. Dipersio, J. F. Liesveld, J. L. McCarty, J. M. Rowley, S. D. Vesole, D. H. Badel, K. Calandra, G. (2005). The use of AMD3100 plus G-CSF for autologous hematopoietic progenitor cell mobilization is superior to G-CSF alone. Blood 106, 1867–1874. Abstract DOI
- Foudi, A. Jarrier, P. Zhang, Y. Wittner, M. Geay, J. F. Lecluse, Y. Nagasawa, T. Vainchenker, W. Louache, F. (2006). Reduced retention of radioprotective hematopoietic cells within the bone marrow microenvironment in CXCR4-/- chimeric mice. Blood 107, 2243–2251. Abstract DOI
- Fraser, S. T. Ogawa, M. Yu, R. T. Nishikawa, S. Yoder, M. C. Nishikawa, S. (2002). Definitive hematopoietic commitment within the embryonic vascular endothelial-cadherin(+) population. Exp Hematol 30, 1070–1078. Article Abstract DOI
- Frenette, P. S. Mayadas, T. N. H. R. Hynes, R. O. Wagner, D. D. (1996). Susceptibility to infection and altered hematopoiesis in mice deficient in both P-and E-selectins. Cell 84, 563–574.
- Frenette, P. S. Subbarao, S. Mazo, I. B. von Andrian, U. H. Wagner, D. D. (1998). Endothelial selectins and vascular cell adhesion molecule-1 promote hematopoietic progenitor homing to bone marrow. Proc Natl Acad Sci U S A 95, 14423–14428. Abstract DOI
- Frenette, P. S. Weiss, L. (2000). Sulfated glycans induce rapid hematopoietic progenitor cell mobilization: evidence for selectin-dependent and independent mechanisms. Blood 96, 2460–2468. Abstract Abstract
- Fukuda, S. Broxmeyer, H. E. Pelus, L. M. (2005). Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha(CXCL12)/CXCR4 axis. Blood 105, 3117–3126. Abstract DOI
- Garcia-Moruja, C. Alonso-Lobo, J. M. Rueda, P. Torres, C. Gonzalez, N. Bermejo, M. Luque, F. Arenzana-Seisdedos, F. Alcami, J. Caruz, A. (2005). Functional characterization of SDF-1 proximal promoter. J Mol Biol 348, 43–62. Abstract DOI
- Gekas, C. Dieterlen-Lievre, F. Orkin, S. H. Mikkola, H. K. (2005). The placenta is a niche for hematopoietic stem cells. Dev Cell 8, 365–375. Abstract DOI
- Godin, I. Cumano, A. (2002). The hare and the tortoise: an embryonic haematopoietic race. Nat Rev Immunol 2, 593–604. Abstract Abstract
- Gong, J. K. (1978). Endosteal marrow: a rich source of hematopoietic stem cells. Science 199, 1443–1445. Abstract DOI
- Goodman, J. W. Hodgson, G. S. (1962). Evidence for stem cells in the peripheral blood of mice. Blood 19, 702–714. Abstract Abstract
- Gurney, A. L. Carver-Moore, K. de Sauvage, F. J. Moore, M. W. (1994). Thrombocytopenia in c-mpl-deficient mice. Science 265, 1445–1447. Abstract DOI
- Haug, J. S. He, X. C. Grindley, J. C. Wunderlich, J. P. Gaudenz, K. Ross, J. T. Paulson, A. Wagner, K. P. Xie, Y. Zhu, R. (2008). N-cadherin expression level distinguishes reserved versus primed states of hematopoietic stem cells. Cell Stem Cell 2, 367–379. Abstract DOI
- Haylock, D. N. Nilsson, S. K. (2005). Stem cell regulation by the hematopoietic stem cell niche. Cell Cycle 4, 1353–1355. Abstract Abstract
- Heissig, B. Hattori, K. Dias, S. Friedrich, M. Ferris, B. Hackett, N. R. Crystal, R. G. Besmer, P. Lyden, D. Moore, M. A. (2002). Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell 109, 625–637. Abstract DOI
- Hidalgo, A. Weiss, L. A. Frenette, P. S. (2002). Functional selectin ligands mediating human CD34(+) cell interactions with bone marrow endothelium are enhanced postnatally. J Clin Invest 110, 559–569. Abstract Abstract
- Hidalgo, A. Peired, A. J. Weiss, L. A. Katayama, Y. Frenette, P. S. (2004). The integrin alphaMbeta2 anchors hematopoietic progenitors in the bone marrow during enforced mobilization. Blood 104, 993–1001. Abstract DOI
- Hidalgo, A. Frenette, P. S. (2005). Enforced fucosylation of neonatal CD34+ cells generates selectin ligands that enhance the initial interactions with microvessels but not homing to bone marrow. Blood 105, 567–575. Abstract DOI
- Hodivala-Dilke, K. M. McHugh, K. P. Tsakiris, D. A. Rayburn, H. Crowley, D. Ullman-Cullere, M. Ross, F. P. Coller, B. S. Teitelbaum, S. Hynes, R. O. (1999). Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest 103, 229–238. Article Abstract DOI
- Imbert, A. M. Belaaloui, G. Bardin, F. Tonnelle, C. Lopez, M. Chabannon, C. (2006). CD99 expressed on human mobilized peripheral blood CD34+ cells is involved in transendothelial migration. Blood 108, 2578–2586. Abstract DOI
- Isidori, A. Motta, M. R. Tani, M. Terragna, C. Zinzani, P. Curti, A. Rizzi, S. Taioli, S. Giudice, V. D’Addio, A. (2007). Positive selection and transplantation of autologous highly purified CD133(+) stem cells in resistant/relapsed chronic lymphocytic leukemia patients results in rapid hematopoietic reconstitution without an adequate leukemic cell purging. Biol Blood Marrow Transplant 13, 1224–1232. Abstract DOI
- Jacobson, L. O. Marks, E. K. Robson, M. J. Gaston, E. Zirkle, R. E. (1949). The effect of spleen protection on mortality following X-radiation. J Lab Clin Med 34, 1538–1543.
- Johnson, G. R. Moore, M. A. (1975). Role of stem cell migration in initiation of mouse foetal liver haemopoiesis. Nature 258, 726–728. Abstract DOI
- Jung, Y. Wang, J. Schneider, A. Sun, Y. X. Koh-Paige, A. J. Osman, N. I. McCauley, L. K. Taichman, R. S. (2006). Regulation of SDF-1 (CXCL12) production by osteoblasts; a possible mechanism for stem cell homing. Bone 38, 497–508. Abstract DOI
- Katayama, Y. Hidalgo, A. Furie, B. C. Vestweber, D. Furie, B. Frenette, P. S. (2003). PSGL-1 participates in E-selectin-mediated progenitor homing to bone marrow: evidence for cooperation between E-selectin ligands and alpha4 integrin. Blood 102, 2060–2067. Abstract DOI
- Katayama, Y. Hidalgo, A. Peired, A. Frenette, P. S. (2004). Integrin alpha4beta7 and its counterreceptor MAdCAM-1 contribute to hematopoietic progenitor recruitment into bone marrow following transplantation. Blood 104, 2020–2026. Abstract DOI
- Katayama, Y. Hidalgo, A. Chang, J. Peired, A. Frenette, P. S. (2005). CD44 is a physiological E-selectin ligand on neutrophils. J Exp Med 201, 1183–1189. Abstract DOI
- Katayama, Y. Battista, M. Kao, W. M. Hidalgo, A. Peired, A. J. Thomas, S. A. Frenette, P. S. (2006). Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124, 407–421. Abstract DOI
- Kaushansky, K. (1995). Thrombopoietin: the primary regulator of platelet production. Blood 86 , 419–431. Abstract Abstract
- Kiel, M. J. Yilmaz, O. H. Iwashita, T. Yilmaz, O. H. Terhorst, C. Morrison, S. J. (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. Abstract DOI
- Kiel, M. J. He, S. Ashkenazi, R. Gentry, S. N. Teta, M. Kushner, J. A. Jackson, T. L. Morrison, S. J. (2007). Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature 449, 238–242. Abstract DOI
- Kiel, M. J. Radice, G. L. Morrison, S. J. (2007). Lack of evidence that hematopoietic stem cells depend on N-cadherin-mediated adhesion to osteoblasts for their maintenance. Cell Stem Cell 1, 204–217. Abstract DOI
- Kiel, M. J. Morrison, S. J. (2008). Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol 8, 290–301. Abstract DOI
- Kim, C. H. Broxmeyer, H. E. (1998). In vitro behavior of hematopoietic progenitor cells under the influence of chemoattractants: stromal cell-derived factor-1, steel factor, and the bone marrow environment. Blood 91, 100–110. Abstract Abstract
- Kollet, O. Dar, A. Shivtiel, S. Kalinkovich, A. Lapid, K. Sztainberg, Y. Tesio, M. Samstein, R. M. Goichberg, P. Spiegel, A. (2006). Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med 12, 657–664. Abstract DOI
- Kopp, H. G. Avecilla, S. T. Hooper, A. T. Rafii, S. (2005). The bone marrow vascular niche: home of HSC differentiation and mobilization. Physiology (Bethesda) 20, 349–356. Abstract Abstract
- Kopp, H. G. Avecilla, S. T. Hooper, A. T. Shmelkov, S. V. Ramos, C. A. Zhang, F. Rafii, S. (2005). Tie2 activation contributes to hemangiogenic regeneration after myelosuppression. Blood 106, 505–513. Article Abstract DOI
- Labow, M. A. Norton, C. R. Rumberger, J. M. Lombard-Gillooly, K. M. Shuster, D. J. Hubbard, J. Bertko, R. Knaack, P. A. Terry, R. W. Harbison, M. L. (1994). Characterization of E-selectin-deficient mice: Demonstration of overlapping function of the endothelial selectins. Immunity 1, 709–720.
- Levesque, J. P. Takamatsu, Y. Nilsson, S. K. Haylock, D. N. Simmons, P. J. (2001). Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood 98, 1289–1297. Abstract DOI
- Levesque, J. P. Hendy, J. Takamatsu, Y. Williams, B. Winkler, I. G. Simmons, P. J. (2002). Mobilization by either cyclophosphamide or granulocyte colony-stimulating factor transforms the bone marrow into a highly proteolytic environment. Exp Hematol 30, 440–449. Abstract DOI
- Levesque, J. P. Hendy, J. Takamatsu, Y. Simmons, P. J. Bendall, L. J. (2003). Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest 111, 187–196. Abstract Abstract
- Levesque, J. P. Liu, F. Simmons, P. J. Betsuyaku, T. Senior, R. M. Pham, C. Link, D. C. (2004). Characterization of hematopoietic progenitor mobilization in protease-deficient mice. Blood 104, 65–72. Abstract DOI
- Li, W. Johnson, S. A. Shelley, W. C. Yoder, M. C. (2004). Hematopoietic stem cell repopulating ability can be maintained in vitro by some primary endothelial cells. Exp Hematol 32, 1226–1237. Abstract DOI
- Liles, W. C. Broxmeyer, H. E. Rodger, E. Wood, B. Hubel, K. Cooper, S. Hangoc, G. Bridger, G. J. Henson, G. W. Calandra, G. Dale, D. C. (2003). Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood 102, 2728–2730. Abstract DOI
- Liu, F. Poursine-Laurent, J. Link, D. C. (1997). The granulocyte colony-stimulating factor receptor is required for the mobilization of murine hematopoietic progenitors into peripheral blood by cyclophosphamide or interleukin-8 but not flt-3 ligand. Blood 90, 2522–2528. Abstract Abstract
- Liu, F. Poursine-Laurent, J. Link, D. C. (2000). Expression of the G-CSF receptor on hematopoietic progenitor cells is not required for their mobilization by G-CSF. Blood 95, 3025–3031. Abstract Abstract
- Lord, B. I. Testa, N. G. Hendry, J. H. (1975). The relative spatial distributions of CFUs and CFUc in the normal mouse femur. Blood 46, 65–72. Abstract Abstract
- Lorenz, E. Uphoff, D. Reid, T. R. Shelton, E. (1951). Modification of irradiation injury in mice and guinea pigs by bone marrow injections. J Natl Cancer Inst 12, 197–201.
- Ma, Q. Jones, D. Borghesani, P. R. Segal, R. A. Nagasawa, T. Kishimoto, T. Bronson, R. T. Springer, T. A. (1998). Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci U S A 95, 9448–9453. Abstract DOI
- Ma, Q. Jones, D. Springer, T. A. (1999). The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity 10, 463–471. Abstract DOI
- Martinez-Agosto, J. A. Mikkola, H. K. Hartenstein, V. Banerjee, U. (2007). The hematopoietic stem cell and its niche: a comparative view. Genes Dev 21, 3044–3060. Abstract DOI
- Massberg, S. Schaerli, P. Knezevic-Maramica, I. Kollnberger, M. Tubo, N. Moseman, E. A. Huff, I. V. Junt, T. Wagers, A. J. Mazo, I. B. von Andrian, U. H. (2007). Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell 131, 994–1008. Abstract DOI
- Matsui, Y. Zsebo, K. M. Hogan, B. L. (1990). Embryonic expression of a haematopoietic growth factor encoded by the Sl locus and the ligand for c-kit. Nature 347, 667–669. Abstract DOI
- Mazo, I. B. Gutierrez-Ramos, J. C. Frenette, P. S. Hynes, R. O. Wagner, D. D. von Andrian, U. H. (1998). Hematopoietic progenitor cell rolling in bone marrow microvessels: parallel contributions by endothelial selectins and vascular cell adhesion molecule 1. J Exp Med 188, 465–474. Abstract DOI
- McCredie, K. B. Hersh, E. M. Freireich, E. J. (1971). Cells capable of colony formation in the peripheral blood of man. Science 171, 293–294. Abstract DOI
- McGrath, K. E. Koniski, A. D. Maltby, K. M. McGann, J. K. Palis, J. (1999). Embryonic expression and function of the chemokine SDF-1 and its receptor, CXCR4. Dev Biol 213, 442–456. Abstract DOI
- McGrath, K. E. Koniski, A. D. Malik, J. Palis, J. (2003). Circulation is established in a stepwise pattern in the mammalian embryo. Blood 101, 1669–1676. Abstract DOI
- McKinney-Freeman, S. L. Jackson, K. A. Camargo, F. D. Ferrari, G. Mavilio, F. Goodell, M. A. (2002). Muscle-derived hematopoietic stem cells are hematopoietic in origin. Proc Natl Acad Sci U S A 99, 1341–1346. Abstract DOI
- Mendes, S. C. Robin, C. Dzierzak, E. (2005). Mesenchymal progenitor cells localize within hematopoietic sites throughout ontogeny. Development 132, 1127–1136. Abstract DOI
- Mendez-Ferrer, S. Lucas, D. Battista, M. Frenette, P. S. (2008). Haematopoietic stem cell release is regulated by circadian oscillations. Nature. Abstract Abstract
- Meuer, K. Pitzer, C. Teismann, P. Kruger, C. Goricke, B. Laage, R. Lingor, P. Peters, K. Schlachetzki, J. C. Kobayashi, K. (2006). Granulocyte-colony stimulating factor is neuroprotective in a model of Parkinson's disease. J Neurochem 97, 675–686. Abstract DOI
- Mikkola, H. K. Orkin, S. H. (2006). The journey of developing hematopoietic stem cells. Development 133, 3733–3744. Abstract DOI
- Minasi, M. G. Riminucci, M. De Angelis, L. Borello, U. Berarducci, B. Innocenzi, A. Caprioli, A. Sirabella, D. Baiocchi, M. De Maria, R. (2002). The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 129, 2773–2783. Abstract DOI
- Miyake, K. Medina, K. Ishihara, K. Kimoto, M. Auerback, R. Kinkade, P. W. (1991). A VCAM-like adhesion molecule on murine bone marrow stromal cells mediates binding of lymphocyte precursors in culture. J Cell Biol 114, 557–565.
- Molineux, G. Pojda, Z. Hampson, I. N. Lord, B. I. Dexter, T. M. (1990). Transplantation potential of peripheral blood stem cells induced by granulocyte colony-stimulating factor. Blood 76, 2153–2158. Abstract Abstract
- Moore, M. A. Metcalf, D. (1970). Ontogeny of the haemopoietic system: yolk sac origin of in vivo and in vitro colony forming cells in the developing mouse embryo. Br J Haematol 18, 279–296. Abstract DOI
- Morrison, S. J. Wandycz, A. M. Hemmati, H. D. Wright, D. E. Weissman, I. L. (1997). Identification of a lineage of multipotent hematopoietic progenitors. Development 124 , 1929–1939. Abstract Abstract
- Morrison, S. J. Spradling, A. C. (2008). Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 132, 598–611. Abstract DOI
- Murayama, E. Kissa, K. Zapata, A. Mordelet, E. Briolat, V. Lin, H. F. Handin, R. I. Herbomel, P. (2006). Tracing hematopoietic precursor migration to successive hematopoietic organs during zebrafish development. Immunity 25, 963–975. Abstract DOI
- Nagasawa, T. Hirota, S. Tachibana, K. Takakura, N. Nishikawa, S. Kitamura, Y. Yoshida, N. Kikutani, H. Kishimoto, T. (1996). Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382, 635–638. Abstract DOI
- Nie, Y. Han, Y. C. Zou, Y. R. (2008). CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. Abstract Abstract
- Nilsson, S. K. Johnston, H. M. Coverdale, J. A. (2001). Spatial localization of transplanted hemopoietic stem cells: inferences for the localization of stem cell niches. Blood 97, 2293–2299. Abstract DOI
- Nilsson, S. K. Johnston, H. M. Whitty, G. A. Williams, B. Webb, R. J. Denhardt, D. T. Bertoncello, I. Bendall, L. J. Simmons, P. J. Haylock, D. N. (2005). Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 106, 1232–1239. Abstract DOI
- Ninomiya, M. Abe, A. Katsumi, A. Xu, J. Ito, M. Arai, F. Suda, T. Ito, M. Kiyoi, H. Kinoshita, T. Naoe, T. (2007). Homing, proliferation and survival sites of human leukemia cells in vivo in immunodeficient mice. Leukemia 21, 136–142. Abstract DOI
- Okumura, N. Tsuji, K. Ebihara, Y. Tanaka, I. Sawai, N. Koike, K. Komiyama, A. Nakahata, T. (1996). Chemotactic and chemokinetic activities of stem cell factor on murine hematopoietic progenitor cells. Blood 87, 4100–4108. Abstract Abstract
- Orkin, S. H. Zon, L. I. (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. Abstract DOI
- Ottersbach, K. Dzierzak, E. (2005). The murine placenta contains hematopoietic stem cells within the vascular labyrinth region. Dev Cell 8, 377–387. Abstract DOI
- Palis, J. Robertson, S. Kennedy, M. Wall, C. Keller, G. (1999). Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. 126, 5073–5084. Abstract Abstract
- Papayannopoulou, T. Craddock, C. Nakamoto, B. Priestley, G. V. Wolf, N. S. (1995). The VLA4/VCAM-1 adhesion pathway defines contrasting mechanisms of lodgement of transplanted murine hemopoietic progenitors between bone marrow and spleen. Proc Natl Acad Sci U S A 92, 9647–9651. Abstract DOI
- Papayannopoulou, T. Priestley, G. V. Nakamoto, B. Zafiropoulos, V. Scott, L. M. (2001). Molecular pathways in bone marrow homing: dominant role of alpha(4)beta(1) over beta(2)-integrins and selectins. Blood 98, 2403–2411. Abstract DOI
- Papayannopoulou, T. Priestley, G. V. Bonig, H. Nakamoto, B. (2003). The role of G-protein signaling in hematopoietic stem/progenitor cell mobilization. Blood 101, 4739–4747. Abstract DOI
- Parmar, K. Mauch, P. Vergilio, J. A. Sackstein, R. Down, J. D. (2007). Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A 104, 5431–5436. Abstract DOI
- Peled, A. Petit, I. Kollet, O. Magid, M. Ponomaryov, T. Byk, T. Nagler, A. Ben-Hur, H. Many, A. Shultz, L. (1999). Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 283, 845–848. Abstract DOI
- Peled, A. Kollet, O. Ponomaryov, T. Petit, I. Franitza, S. Grabovsky, V. Slav, M. M. Nagler, A. Lider, O. Alon, R. (2000). The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood 95, 3289–3296. Abstract Abstract
- Pelus, L. M. Bian, H. King, A. G. Fukuda, S. (2004). Neutrophil-derived MMP-9 mediates synergistic mobilization of hematopoietic stem and progenitor cells by the combination of G-CSF and the chemokines GRObeta/CXCL2 and GRObetaT/CXCL2delta4. Blood 103, 110–119. Abstract DOI
- Pelus, L. M. Bian, H. Fukuda, S. Wong, D. Merzouk, A. Salari, H. (2005). The CXCR4 agonist peptide, CTCE-0021, rapidly mobilizes polymorphonuclear neutrophils and hematopoietic progenitor cells into peripheral blood and synergizes with granulocyte colony-stimulating factor. Exp Hematol 33, 295–307. Abstract DOI
- Perez, L. E. Alpdogan, O. Shieh, J. H. Wong, D. Merzouk, A. Salari, H. O’Reilly, R. J. van den Brink, M. R. Moore, M. A. (2004). Increased plasma levels of stromal-derived factor-1 (SDF-1/CXCL12) enhance human thrombopoiesis and mobilize human colony-forming cells (CFC) in NOD/SCID mice. Exp Hematol 32, 300–307. Abstract DOI
- Petit, I. Szyper-Kravitz, M. Nagler, A. Lahav, M. Peled, A. Habler, L. Ponomaryov, T. Taichman, R. S. Arenzana-Seisdedos, F. Fujii, N. (2002). G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol 3, 687–694. Abstract DOI
- Petit, I. Goichberg, P. Spiegel, A. Peled, A. Brodie, C. Seger, R. Nagler, A. Alon, R. Lapidot, T. (2005). Atypical PKC-zeta regulates SDF-1-mediated migration and development of human CD34+ progenitor cells. J Clin Invest 115, 168–176. Abstract Abstract
- Potocnik, A. J. Brakebusch, C. Fassler, R. (2000). Fetal and adult hematopoietic stem cells require beta1 integrin function for colonizing fetal liver, spleen, and bone marrow. Immunity 12, 653–663. Abstract DOI
- Qian, H. Tryggvason, K. Jacobsen, S. E. Ekblom, M. (2006). Contribution of alpha6 integrins to hematopoietic stem and progenitor cell homing to bone marrow and collaboration with alpha4 integrins. Blood 107, 3503–3510. Abstract DOI
- Qian, H. Georges-Labouesse, E. Nystrom, A. Domogatskaya, A. Tryggvason, K. Jacobsen, S. E. Ekblom, M. (2007). Distinct roles of integrins alpha6 and alpha4 in homing of fetal liver hematopoietic stem and progenitor cells. Blood 110, 2399–2407. Abstract DOI
- Ratajczak, J. Reca, R. Kucia, M. Majka, M. Allendorf, D. J. Baran, J. T. Janowska-Wieczorek, A. Wetsel, R. A. Ross, G. D. Ratajczak, M. Z. (2004). Mobilization studies in mice deficient in either C3 or C3a receptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood 103, 2071–2078. Abstract DOI
- Rhodes, K. E. Gekas, C. Wang, Y. Lux, C. T. Francis, C. S. Chan, D. N. Conway, S. Orkin, S. H. Yoder, M. C. Mikkola, H. K. (2008). The emergence of hematopoietic stem cells is initiated in the placental vasculature in the absence of circulation. Cell Stem Cell 2, 252–263. Abstract DOI
- Richman, C. M. Weiner, R. S. Yankee, R. A. (1976). Increase in circulating stem cells following chemotherapy in man. Blood 47, 1031–1039. Abstract Abstract
- Sacchetti, B. Funari, A. Michienzi, S. Di Cesare, S. Piersanti, S. Saggio, I. Tagliafico, E. Ferrari, S. Robey, P. G. Riminucci, M. Bianco, P. (2007). Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131, 324–336. Abstract DOI
- Sackstein, R. Merzaban, J. S. Cain, D. W. Dagia, N. M. Spencer, J. A. Lin, C. P. Wohlgemuth, R. (2008). Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med 14, 181–187. Abstract DOI
- Samokhvalov, I. M. Samokhvalova, N. I. Nishikawa, S. (2007). Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 446, 1056–1061. Abstract DOI
- Schneider, A. Kruger, C. Steigleder, T. Weber, D. Pitzer, C. Laage, R. Aronowski, J. Maurer, M. H. Gassler, N. Mier, W. (2005). The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J Clin Invest 115, 2083–2098. Abstract DOI
- Schweitzer, K. M. Drager, A. M. van der Valk, P. Thijsen, S. F. T. Zevenbergen, A. Theijsmeijer, A. P. van der Schoot, C. E. Langenhuijsen, M. M. A. C. (1996). Constitutive expression of E-selectin and vascular cell adhesion molecule-1 on endothelial cells of hematopoietic tissues. Am J Pathol 148, 165–175.
- Scott, L. M. Priestley, G. V. Papayannopoulou, T. (2003). Deletion of alpha4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Mol Cell Biol 23, 9349–9360. Abstract DOI
- Semerad, C. L. Liu, F. Gregory, A. D. Stumpf, K. Link, D. C. (2002). G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity 17, 413–423. Abstract DOI
- Shen, H. Cheng, T. Olszak, I. Garcia-Zepeda, E. Lu, Z. Herrmann, S. Fallon, R. Luster, A. D. Scadden, D. T. (2001). CXCR-4 desensitization is associated with tissue localization of hemopoietic progenitor cells. J Immunol 166, 5027–5033. Abstract Abstract
- Sipkins, D. A. Wei, X. Wu, J. W. Runnels, J. M. Cote, D. Means, T. K. Luster, A. D. Scadden, D. T. Lin, C. P. (2005). In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435, 969–973. Abstract DOI
- Soni, S. Bala, S. Gwynn, B. Sahr, K. E. Peters, L. L. Hanspal, M. (2006). Absence of erythroblast macrophage protein (Emp) leads to failure of erythroblast nuclear extrusion. J Biol Chem 281, 20181–20189. Abstract DOI
- Stier, S. Ko, Y. Forkert, R. Lutz, C. Neuhaus, T. Grunewald, E. Cheng, T. Dombkowski, D. Calvi, L. M. Rittling, S. R. Scadden, D. T. (2005). Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med 201, 1781–1791. Abstract DOI
- Sugiyama, T. Kohara, H. Noda, M. Nagasawa, T. (2006). Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988. Abstract DOI
- Sweeney, E. A. Lortat-Jacob, H. Priestley, G. V. Nakamoto, B. Papayannopoulou, T. (2002). Sulfated polysaccharides increase plasma levels of SDF-1 in monkeys and mice: involvement in mobilization of stem/progenitor cells. Blood 99, 44–51. Abstract DOI
- Taichman, R. S. Reilly, M. J. Emerson, S. G. (1996). Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood 87, 518–524. Abstract Abstract
- Takamatsu, Y. Simmons, P. J. Moore, R. J. Morris, H. A. To, L. B. Levesque, J. P. (1998). Osteoclast-mediated bone resorption is stimulated during short-term administration of granulocyte colony-stimulating factor but is not responsible for hematopoietic progenitor cell mobilization. Blood 92, 3465–3473. Abstract Abstract
- Taoudi, S. Morrison, A. M. Inoue, H. Gribi, R. Ure, J. Medvinsky, A. (2005). Progressive divergence of definitive haematopoietic stem cells from the endothelial compartment does not depend on contact with the foetal liver. 132, 4179–4191. Abstract Abstract
- Tavassoli, M. Hardy, C. L. (1990). Molecular basis of homing of intravenously transplanted stem cells to the marrow. Blood 76, 1059–1070. Abstract Abstract
- Thomas, E. D. (1995). History, current results, and research in marrow transplantation. Persp Biol Med 38, 230–237.
- Tokoyoda, K. Egawa, T. Sugiyama, T. Choi, B. I. Nagasawa, T. (2004). Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 20, 707–718. Abstract DOI
- Ulyanova, T. Scott, L. M. Priestley, G. V. Jiang, Y. Nakamoto, B. Koni, P. A. Papayannopoulou, T. (2005). VCAM-1 expression in adult hematopoietic and nonhematopoietic cells is controlled by tissue-inductive signals and reflects their developmental origin. Blood 106, 86–94. Abstract DOI
- Vermeulen, M. Le Pesteur, F. Gagnerault, M. C. Mary, J. Y. Sainteny, F. Lepault, F. (1998). Role of adhesion molecules in the homing and mobilization of murine hematopoietic stem and progenitor cells. Blood 92, 894–900. Abstract Abstract
- Visnjic, D. Kalajzic, Z. Rowe, D. W. Katavic, V. Lorenzo, J. Aguila, H. L. (2004). Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 103, 3258–3264. Abstract DOI
- Warren, S. Chute, R. N. Farrington, E. M. (1960). Protection of the hematopoietic system by parabiosis. Lab Invest 9, 191–198. Abstract Abstract
- Wilson, A. Murphy, M. J. Oskarsson, T. Kaloulis, K. Bettess, M. D. Oser, G. M. Pasche, A. C. Knabenhans, C. Macdonald, H. R. Trumpp, A. (2004). c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 18, 2747–2763. Abstract DOI
- Winkler, I. G. Hendy, J. Coughlin, P. Horvath, A. Levesque, J. P. (2005). Serine protease inhibitors serpina1 and serpina3 are down-regulated in bone marrow during hematopoietic progenitor mobilization. J Exp Med 201, 1077–1088. Abstract DOI
- Wright, D. E. Wagers, A. J. Gulati, A. P. Johnson, F. L. Weissman, I. L. (2001). Physiological migration of hematopoietic stem and progenitor cells. Science 294, 1933–1936. Abstract DOI
- Xia, L. McDaniel, J. M. Yago, T. Doeden, A. McEver, R. P. (2004). Surface fucosylation of human cord blood cells augments binding to P-selectin and E-selectin and enhances engraftment in bone marrow. Blood 104, 3091–3096. Abstract DOI
- Yahata, T. Ando, K. Sato, T. Miyatake, H. Nakamura, Y. Muguruma, Y. Kato, S. Hotta, T. (2003). A highly sensitive strategy for SCID-repopulating cell assay by direct injection of primitive human hematopoietic cells into NOD/SCID mice bone marrow. Blood 101, 2905–2913. Abstract DOI
- Yong, K. L. Watts, M. Shaun Thomas, N. Sullivan, A. Ings, S. Linch, D. C. (1998). Transmigration of CD34+ cells across specialized and nonspecialized endothelium requires prior activation by growth factors and is mediated by PECAM-1 (CD31). Blood 91, 1196–1205. Abstract Abstract
- Zhang, J. Niu, C. Ye, L. Huang, H. He, X. Tong, W. G. Ross, J. Haug, J. Johnson, T. Feng, J. Q. (2003). Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425, 836–841. Abstract DOI
- Zhang, J. Grindley, J. C. Yin, T. Jayasinghe, S. He, X. C. Ross, J. T. Haug, J. S. Rupp, D. Porter-Westpfahl, K. S. Wiedemann, L. M. (2006). PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 441, 518–522. Abstract DOI
- Zhang, X. Y. Rodaway, A. R. (2007). SCL-GFP transgenic zebrafish: in vivo imaging of blood and endothelial development and identification of the initial site of definitive hematopoiesis. Dev Biol 307, 179–194. Abstract DOI
- Zhu, J. Garrett, R. Jung, Y. Zhang, Y. Kim, N. Wang, J. Joe, G. J. Hexner, E. Choi, Y. Taichman, R. S. Emerson, S. G. (2007). Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood 109, 3706–3712. Abstract DOI