
1. Introduction
1.1. Regulation of gene expression
Transcription regulation constitutes a major component of biological activities in the cell. A key function of transcriptional activity is the control of gene expression necessary for the maintenance of a cellular state. In metazoan, all cell types, with the exception of mature germ cells, contain the same genetic information within the species. Yet, each cell type expresses a unique subset of all genes encoded. Differential gene expression can be controlled at many levels. One of the foremost steps occurs at the first stage of expression: transcription. Coding and non-coding genes contain promoter and enhancer sequences that are bound by transcription activators and repressors – transcription factors (TFs; Struhl, 1995). A large proportion of TFs are DNA-binding proteins that localize to cis-regulatory DNA elements located upstream or downstream of their target genes. The levels and activities of TFs determine to a large part the extent of transcription. Hence, the active quantity of these regulators in turn must be tightly regulated. The regulatory mechanisms of TFs are diversified by the combinatorial action of upstream regulatory genes, post-translational modifications (Hill and Treisman, 1995), ligand-dependant activation for nuclear hormone receptors (Beato et al., 1995; Mangelsdorf et al., 1995), and protein-protein interactions (Swillens and Pirson, 1994; Tsai and O’Malley, 1994). Two classes of TFs can be distinguished in the eukaryotes. General transcription factors (GTFs) bind to a core promoter usually close to the transcription initiation site, together with the assembly of the pre-initiation complex, while sequence-specific TFs bind regulatory DNA elements at varying distances away from the transcription start site. The occupancy of transcription activators on a promoter recruits the transcription initiation machinery which consists of RNA polymerases and more than 50 other components (Green, 2000; Johnson et al., 2001; Myer and Young, 1998; Roeder, 1996). This is followed by promoter clearance, elongation, and the termination of transcription. Other components of the post-transcription machinery are also involved in augmenting the overall levels of gene transcripts in the cell. These include regulated mRNA decay which is achieved through the interactions between trans-acting factors with the mRNA's structural components, the sequence dependent mRNA stability, and the association of mRNA with general or mRNA-specific RNA-binding proteins (Guhaniyogi and Brewer, 2001). Therefore, transcription regulation, coupled with post-transcriptional modifications, help determine the base-line of differential gene expression profile that confers cellular identity.
1.2. A stem cell window into transcription factor function
Stem cells are well-suited for the study of transcription regulation and TF function. As these remarkable cells are characterized by their ability to self-renew as well as differentiate into functional cell types, they respond to external stimuli and intracellular perturbations that are observable and detectable. During the course of animal development, cells transit from a pluripotent state to one of many committed lineages. Several developmental stage-specific stem or progenitor cells have been detected and isolated from the mammalian embryo, as well as from post-natal and adult tissues. These include lineage-committed multipotent stem or progenitor cells such as hematopoietic stem cells (HSCs), neural progenitor cells (NPCs), epithelial stem cells and mesenchymal stem cells. Of particular interest are cells of the embryonic inner cell mass (ICM) and their derivatives, the embryonic stem cells (ESCs), which are capable of giving rise to all subsequent cell types of the fetus. This makes ESCs highly promising in regenerative medicine. An important requirement for directing differentiation of stem cells is to first understand how the undifferentiated ‘stemness’ state is established and maintained. This would give us clues on how to isolate and expand stem cell population, as well as control their cell fate specification either chemically or genetically.
At the heart of cellular differentiation programs lies a plethora of highly regulated TFs that enable the alteration of cell states in response to extracellular signaling cues. For many decades, the study of TFs as regulators of downstream effectors has been limited in scope and comprehensiveness. Deciphering the sequential order in which top-tier regulators are activated and identifying what are the extensive repertoire of target genes has been difficult. Different approaches have been employed to address these issues. During cell fate specification, the activation of a hierarchy of TFs has been inferred from the analyses of mutant animals. This is best exemplified in gene knockouts of TFs involved in specific tissue development such as myogenesis (Edmondson and Olson, 1989; Hasty et al., 1993; Rudnicki et al., 1993), hematopoiesis (Huang et al., 2008; Kim et al., 2007; Orkin and Zon, 2008; Park et al., 2003) and neurogenesis (Ma et al., 1996). Downstream perturbations arising from the loss of a key TF regulator have provided clues for the identity of regulated target genes. In recent years, the development of microarray technology and sequencing of many vertebrate genomes has facilitated in capturing gene expression profiles on a global scale and the unraveling of transcription regulation hierarchy. One strategy involves perturbing the TF of interest by either over-expression or depletion, followed by analysis of the resultant changes in gene expression profiles (Menssen and Hermeking, 2002; Stein and Liang, 2002; Werner, 2001a; Werner, 2001b), as exemplified in the attempt to identify target genes of the c-Myc proto-oncogene (Menssen and Hermeking, 2002). However, whether differentially expressed genes are directly bound and regulated by the TF, or indirectly affected through the perturbation of intermediate genes, cannot be distinguished. Therefore, alternative approaches are required to map TF binding sites in the genome accurately and efficiently.
1.3. Interrogation of transcription factor binding sites
Chromatin immunoprecipitation (ChIP) allows for the identification of physical interactions between proteins and DNA in the context of chromatin (Orlando and Paro, 1993; Solomon et al., 1988). An overview of the experimental procedure is depicted in Figure 1. ChIP involves chemical cross-linking of DNA-protein interactions in living cells to capture TFs to their cognate binding sites. Cross-linked chromatin is fragmented and protein-DNA complexes can be enriched by immunoaffinity pull-down of the desired protein. DNA-protein cross-links are then reversed, and the enriched DNA can be purified for analyses. When combined with high-throughput detection instrumentations, ChIP can provide a comprehensive view of protein-DNA interactions. Three major strategies have been employed for locating TF occupancy in a genome-wide manner (reviewed in (Massie and Mills, 2008)). Briefly, ChIP-chip involves the amplification of ChIP-DNA followed by hybridization to whole-genome promoters or customized DNA microarrays (Iyer et al., 2001; Ren et al., 2000). The difference in fluorescent signal intensity of ChIP-DNA over control DNA reflects the relative enrichment of the TF on genomic binding sites. ChIP-PET (paired ends di-tags) represents a sequencing-based method for detecting enrichment of ChIP-DNA (Wei et al., 2006). It involves the construction of a concatenated DNA fragment plasmid library which is then sequenced. Mapping both ends of a fragment has the advantage of accurately decoding the identity of a gene fragment, and this method is not constrained by array-based limitations such as probe performance and genome coverage. Recently, the application of ChIP-seq (sequencing) demonstrated considerable advantages over the other methods (Johnson et al., 2007b; Robertson et al., 2007). ChIP-seq involves direct sequencing of ChIP-DNA which has been size-selected and amplified. The method is simple, requires small amounts of ChIP-DNA, has deeper coverage, and is more efficient. Therefore, the availability of these methods has greatly enhanced our understanding of the complexity that governs transcription regulatory networks in higher eukaryotes. In addition, genome-wide ChIP analysis has also been useful for the study of non-TFs that associate with, or are components of, the chromatin. These proteins include modified histones and epigenetic regulators such as Polycomb complexes (Bernstein et al., 2006; Boyer et al., 2006; Guenther et al., 2007; Lee et al., 2006; Mikkelsen et al., 2007; Sadikovic et al., 2008).
Genome-wide TF localization analysis has provided invaluable insights into the functions of regulators involved in cell homeostasis, cell fate transition, immune defense and cancer (Acosta-Alvear et al., 2007; Cao et al., 2006; Carroll et al., 2005; Li et al., 2003; Lim et al., 2007; Lupien et al., 2008; Marson et al., 2007; Odom et al., 2006; Odom et al., 2004; Palomero et al., 2006). We and others have employed this strategy to examine the transcription circuitry of ESCs which is governed by a hierarchy of core factors. In uncovering and reconstructing the key features of this circuitry, we further dissected the significance and contributions of these transcriptional components to ESC function. The spatial and temporal specifications of stem cells in the embryo and adult somatic tissues have raised important questions pertaining to their establishment and maintenance. In particular, the contrasting role of TFs either as regulators in stem cell maintenance or directing differentiation is of immense interest. Thus, mapping the stem cell transcription network can provide useful clues into their manipulation. Here, we discuss key findings into the deconstruction of molecular circuitries governing stem cell function, with an emphasis on ESCs. Knowledge drawn for these studies has further led to the synthesis of key concepts in helping us understand the complexity of gene regulatory network in stem cells.
2. Key regulators of pluripotency
During development, the formation of the ICM in the mammalian blastocyst is a major landmark that represents the specification of pluripotent cells. ESCs have been derived from human and mouse blastocysts (Evans and Kaufman, 1981; Reubinoff et al., 2000; Thomson et al., 1998), and are similar in many developmental aspects. Although differences exist in their culture and maintenance, many molecular determinants governing their undifferentiated states are shared. Homeodomain TFs are evolutionary conserved and participate in cell fate determination in many organisms (Hombria and Lovegrove, 2003). The POU (Pit/Oct/Unc) TF, Oct4, has emerged as a dominant regulator in ESCs (Scholer et al., 1990a; Scholer et al., 1990b). It is exclusively expressed in blastomeres, pre-implantation embryos, primodial germ cells, the ICM and a narrow range of cultured cell types (Nichols et al., 1998; Pesce et al., 1998; Rosner et al., 1990; Scholer et al., 1990a; Yeom et al., 1996). As a TF, Oct4 can activate or repress gene expression (Botquin et al., 1998; Nichols et al., 1998; Pesce and Scholer, 2001; Scholer et al., 1991), and interact with other molecules such as the HMG-box TF, Sox2, in pluripotent cells (Scholer et al., 1991; Yuan et al., 1995). Genetic studies have confirmed the importance of Oct4 during embryonic development and in maintaining pluripotent ESCs (Nichols et al., 1998; Palmieri et al., 1994). Nanog, another homeodomain-containing TF expressed abundantly in ESCs, primodial germ cells and embryonal carcinoma cell lines, is also necessary for ESC function (Chambers et al., 2003; Mitsui et al., 2003; Ramalho-Santos et al., 2002; Wang et al., 2003). In the mouse embryo, Nanog first appears in the compacted morula, becomes localized to the ICM, and is finally restricted to the epiblast (Chambers et al., 2003). In Nanog−/− embryos, there is a lack of epiblast and primitive ectoderm formations, indicative of the absolute requirement for Nanog; and Nanog−/− ESCs manifest a reduction of pluripotency and re-specification to the extra-embryonic endoderm lineages (Mitsui et al., 2003). Strikingly, a recent observation has clarified the role of Nanog further. While Nanog is necessary for inner cell mass formation and primodial germ cell specification, it appears dispensable in ESCs since Nanog−/− ESCs persist in culture although these cells are more prone to differentiation (Chambers et al., 2007).
A host of other TFs also appear crucial for ESC function. The neuronal repressor REST is highly abundant in mouse ESCs (Ballas et al., 2005), and its depletion either through RNA interference (RNAi) or heterozygous deletion leads to a loss of self-renewal (Singh et al., 2008). REST is a major transcriptional repressor of neurogenesis, and the activation of REST-regulated genes can convert neural progenitor cells to neuronal phenotypes (Ballas and Mandel, 2005; Coulson, 2005; Su et al., 2004). But, the manner in which it regulates gene expression in ESCs is unclear. Another regulator, Sall4, is an important activator of Oct4, and its loss leads to cell fate re-specification into trophoblast stem cells (Elling et al., 2006; Sakaki-Yumoto et al., 2006; Zhang et al., 2006). This effect is recapitulated during pre-implantation development where down-regulation of Sall4 causes mis-expression of the trophoblast stem cell determinant, Cdx2, in the ICM (Zhang et al., 2006). The use of integrated approaches combining transcriptome profiling, genome-wide ChIP analyses, as well as large-scale gain- or loss-of-function has further led to the identification of an increasing number of developmentally important TFs. In these studies, Tcl1, Tbx3 and Esrrb have been identified as novel regulators in ESCs (Ivanova et al., 2006; Loh et al., 2006). However, the precise manner in which these genes exert their regulatory effects has not yet been elucidated.
3. The transcriptional landscape in ESCs
Initial studies into transcriptional regulation by Oct4, Sox2, Nanog and Sall4 have revealed a small number of genes bound by these factors (Bohm et al., 2007; Chew et al., 2005a; Nishimoto et al., 1999; Rodda et al., 2005a; Tokuzawa et al., 2003; Tomioka et al., 2002; Wu et al., 2006; Zhang et al., 2006). These limited observations are simplistic, and do not provide a comprehensive understanding into the complex framework of transcription hierarchy where regulators tend to exert far-reaching effects on a vast array of genes, as observed from yeast studies (Iyer et al., 2001; Lee et al., 2002; Ren et al., 2000). In animals as diverse as C. elegans and Drosophila to Xenopus, mouse and man, activating and inhibiting signals establish a dynamic cascade of coordinated gene expression in response to extrinsic signals and intrinsic programming. The integration of these instructions occurs in the nucleus through combinations of signal-activated and tissue-restricted TFs binding to, and controlling, cis-regulatory modules of genes. With the complexities arising from such intricate interactions, the ESC model, which shows a remarkable sensitivity to genetic perturbations, thus represents a suitable system for probing TF function and regulatory dynamics.
3.1. Auto-regulation and feedback loops maintain gene expression
One emergent theme arising from transcription regulation in ESCs is the revelation that a core set of factors act together in a tightly regulated framework to control similar as well as divergent downstream genes (see Table 1). Auto-regulation by top-tier TFs is a key feature. The pluripotent molecules Oct4, Sox2, Nanog and Sall4 auto-regulate through binding to their own promoters (Boyer et al., 2005; Chew et al., 2005b; Loh et al., 2006; Rodda et al., 2005b; Tomioka et al., 2002; see Figure 2A). On its own, each factor maintains its expression that is individually required for ESC function. Collectively, all four factors regulate one another through the interconnectivity of positive feedback loops (see Table 1). The generation of these feedback circuits reinforces the transcription activity of each factor, and maintains appropriate levels of the necessary factors.
Table 1.
Models of transcription regulatory network
Key Concepts |
Remarks |
Model |
|---|---|---|
Auto-regulatory and feedback loop |
Transcription factors (X and Y) auto-regulate by binding to their own promoters. X and Y also bind the promoters of each other in a feedback loop. Auto-regulation and feedback loops are features of ‘master’ regulators in the transcription network of eukaryotes. Provides enhanced stability of gene expression that maintains high levels of both factors. Examples: Oct4, Sox2, Nanog and Sall4 in ESCs; HNF1α, HNF4α and HNF6 in hepatocytes. |
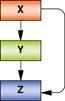 |
Feed-forward loop |
Transcription factor (X) controls another regulator (Y), and both of them regulate downstream target gene (Z). Multiple positive inputs provide consistent activity in regulating a common target, rendering it insensitive to transient changes in individual input. Particularly important in stem cells to maintain either a stable self-renewing or differentiated state. Examples: In ESCs, Nanog regulates Oct4 and Sall4, where Oct4 also controls Sall4; upon differentiation, Sall4 is down-regulated partly owing to the loss of Nanog and Oct4. |
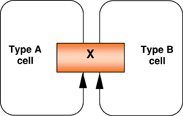 |
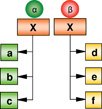
One transcription factor → Multiple target genes |
A regulator (X) controls the expression of many target genes. Dominant key factors control a wide repertoire of genes in a positive or negative manner, which may require the activity of co-factors. The sum of activity of all downstream targets represents the cellular state. Example: REST represses extensive neuronal lineage-associated targets in non-neuronal cells; HNF1α controls hepatocyte biochemistry-related genes in liver; Oct4 activates ESC-associated genes and represses differentiation-associated genes in ESCs. |
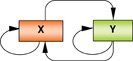 |
Core set of transcription factors → Common target genes |
Transcription factors (X, Y and Z) forms a central regulatory hub. All three factors tend to bind common target genes, in addition to other targets. Core factors are known to regulate a set of common downstream regulators which themselves are crucial for the maintenance of a particular cellular state. Examples: Oct4, Sox2, Nanog regulates common genes in undifferentiated ESCs; MyoD and MyoG maintains the expression of common genes during myogenesis in a temporal manner. |
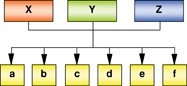 |
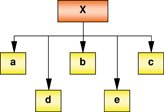
Combinatorial inputs → Transcription output |
Transcription factors comprising of activator (A) and repressor (R) bind to a common target gene. The combinatorial input of activators and repressors determine the transcription output of Y. Output may be binary (On/Off) or graded (High/Low). Repressors may behave as ‘transistors’ which moderate gene expression levels. Example: Regulation of the Oct4 gene activity by the activator Nanog and the repressor Tcf3 in ESCs. |
 |
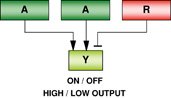
One transcription factor → Distinct circuitries |
The transcription factor (X) may control different and/or overlapping sets of target genes in a cell-type dependent manner. The regulation of dissimilar gene sets is responsible for a varied role in different cell types or cellular contexts. The control of a similar set of genes may suggest a common function for the factor in different cell types. Example: Sall4 maintains lineage-distinct ESCs and XEN cells by binding to different sets of genes; c-Myc regulates similar proliferation-associated genes in ESCs and cancer cells. |
 |
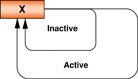
Co-factor dependent recruitment to targets |
The activity and target gene occupancy profile of a transcription factor (X) is dependent on its association with specific co-factors (α or β). Co-factors can mediate recruitment of X to specific target genes that may be motif (DNA sequence) dependent. Example: FoxA1 recruitment to differential targets in breast and prostate cancer cells is mediated by the ERα and AR, respectively. |
 |
Condition-dependent recruitment to targets |
The transcription factor (X) regulates a set of target genes in the native state of the cell. Upon stimuli, the binding dynamics of X may become expanded or altered to include either a larger repertoire of targets relevant for a specific response, or an entirely different set of genes. The expression of co-factors induced by stimulation may be necessary for recruitment to targets. Example: Stimulation of monocytes by LPS expands the binding profile of NF-κB which is partly dependent upon E2F1. |
 |
Recently, the transcription landscape has been extended through the analyses of a greater number of factors associated with ESC pluripotency, as well as those implicated in the reprogramming of somatic cells to induced-pluripotent stem (iPS) cells. The extended repertoire examined reinforces previous observations that auto-regulatory and feedback loops are prevalent features of ESC transcriptional landscape. In one study, the interconnectivity of nine TFs (Oct4, Sox2, Nanog, Dax1, Nac1, Klf4, Zfp281, Rex1 and c-Myc) has been analyzed. Of these regulators, the promoters of Oct4, Sox2, Nanog and Dax1 are bound by at least four factors including itself (Kim et al., 2008), suggestive of a robust positive feedback network. Strikingly, new regulatory foci that include Sall4, Rif1, REST and Dax1, bound by multiple TFs, have also been uncovered (Kim et al., 2008), integrating these hubs to the core circuitry.
Auto-regulation and feedback loops appear to confer several advantages. A core set of factors that maintains its own expression, as well as that of others in the vicinity, enhances the stability of gene expression (Boyer et al., 2005; McAdams and Arkin, 1997). This is especially vital to ESCs since the prolonged loss of any key factor is sufficient to cause irreversible differentiation. Based on empirical evidence that have emerged from biological studies, the computational notion of a ‘bistable’ switch arising from the positive feedback loops comprised of Oct4, Sox2 and Nanog has been proposed (Chickarmane et al., 2006). It is thought that such a mechanism stabilizes the expression levels of all three factors, which through their regulatory control of downstream targets, appears extremely tolerant to parameter change and could counter input noise (Chickarmane et al., 2006; Rao et al., 2002).
3.2. Feed-forward regulation of common target genes
The second observation is the regulation of common target genes by ESC regulators. Oct4, Sox2 and Nanog form feed-forward loops (see Table 1) that involve over 350 protein-coding genes in human ESCs (Boyer et al., 2005). In the mouse counterpart, Oct4 and Nanog occupy approximately 1,000 and 3,000 genomic binding sites respectively (Loh et al., 2006). Many of these Oct4-bound loci also contain Sox2 sites, confirming both factors act in tandem. Significantly, Oct4 and Nanog positively control downstream genes important for maintaining pluripotency and inhibiting differentiation. These included Foxd3, Setdb1, Rif1, Esrrb and n-Myc which help maintain pluripotency (Adams and McLaren, 2004; Cartwright et al., 2005; Dodge et al., 2004; Guo et al., 2002; Hanna et al., 2002; Luo et al., 1997; Mitsunaga et al., 2004). A cascade of other pathways that are connected to self-renewal, genome surveillance and cell fate determination are also implicated (Loh et al., 2006). In addition to their role as activators, Oct4 and Nanog can repress differentiation-associated genes. One key example repressed by Oct4 is Cdx2, necessary for trophectoderm formation (Niwa et al., 2005). Additionally, integrated analysis of a larger set of TFs revealed that target genes co-occupied simultaneously by multiple other factors (Oct4, Sox2, Nanog, Dax1, Nac1 or Klf4), are found to be generally active in ESCs, and repressed upon differentiation; whereas those bound by a single factor alone tend to be inactive or repressed (Kim et al., 2008; see Figure 3A). This suggests multiple inputs may be necessary for a concerted activation signal, possibly through the assembly of activation complexes. For single inputs, these could be associated with co-repression complexes, as revealed by protein network analysis where there exist multiple connections between core factors and repressive chromatin complexes such as NuRD and Polycomb (Wang et al., 2006).
Interestingly, discrete feed-forward loops comprised of divergent target sets bound by different subsets of TFs can be discerned. In a separate study involving the genome-wide mapping of 13 TFs (Oct4, Sox2, Nanog, Klf4, c-, n-MycMyc, n-Myc, Esrrb, Zfx, Tcfcp2l1, Stat3, Smad1, E2f1 and CTCF), it is striking that whereas targets of Oct4, Sox2, Nanog, Stat3 and Smad1 appear more closely associated based on hierarchical clustering, targets of c-Myc, n-Myc and Zfx fall into a separate grouping (Chen et al., 2008; Kim et al., 2008) .The interrogation of chromatin occupancy for a large set of TFs, not limited to promoters, has surprisingly revealed genomic regions, or hotspots, which are co-occupied. If only a few factors were examined, the existence of these hotspots would not have been evident. These nucleoprotein complexes comprised of a combination of TFs binding to enhancer DNA are referred to as enhanceosomes (Thanos and Maniatis, 1995). It is furthermore striking that the densest binding loci occupied by 12 of 13 factors is the previously characterized distal enhancer of Oct4 (Chen et al., 2008; Chew et al., 2005a; see Figure 3B). Several factors in this complex have already been reported to interact with one another (Chew et al., 2005a; Okumura-Nakanishi et al., 2005; Suzuki et al., 2006; Wang and Orkin, 2008). Given the limited understanding of enhancer functions, in-depth analyses of enhanceosomes, or common target loci, could provide valuable insights into key regulatory regions unique to ESCs.
The reliance on feed-forward loops has several advantages. When more than one positive regulator control a common gene, either directly or indirectly through intermediates, the multiple inputs provide consistent activity that render it relatively insensitive to transient changes in individual input strength (Mangan and Alon, 2003; Mangan et al., 2003; see Table 1). When these regulators have either a positive or negative function, the feed-forward loop then behaves as a sensor that can respond rapidly to the balance of the inputs. In that sense, the upstream genetic cis-regulatory elements can be viewed as a ‘transistor’ for receiving and moderating the input signals of the TFs which may carry either an activating or repressive function. This concept of feed-forward loops appears particularly vital to stem cells which must react appropriately to either self-renewal or differentiation cues (Boyer et al., 2005).
Much less is understood about the consequences of having elevated levels of pluripotency factors. As exemplified by Oct4 and Nanog, their elevation at non-physiological levels dramatically alters ESCs. An up-regulation of Oct4 causes ESCs differentiation towards the mesendoderm lineage while an increase in Nanog limits their ability to differentiate (Chambers et al., 2003; Mitsui et al., 2003; Niwa et al., 2000; Tam et al., 2008). Furthermore, the ectopic mis-expression of Oct4 in somatic tissues of adult mice can lead to dysplastic growth in epithelial tissues and block progenitor cell differentiation (Hochedlinger et al., 2005). Thus, it is imperative to understand the fine-tuning of appropriate transcription output by repressors.
3.3. Fine-tuning the transcription output
The negative regulation of pluripotency-associated genes is poorly understood and largely under-appreciated. The tumor suppressor p53, for example, suppresses transcription of Nanog and induces differentiation in response to DNA damage, suggesting its role in maintaining genetic stability (Lin et al., 2005); and, the orphan nuclear receptor GCNF, was reported to repress Oct4 and Nanog during retinoic acid-induced differentiation of ESCs (Gu et al., 2005). Recent studies have further highlighted the role of the Wnt TF, Tcf3, as a potent repressor of Oct4 and Nanog (Pereira et al., 2006; Tam et al., 2008; Yi et al., 2008a). The Wnt signaling cascade is frequently implicated in the establishment of stem cell identity, as well as directing cell fate commitment (Hirabayashi et al., 2004; Huelsken et al., 2001; Lagutin et al., 2003; Merrill et al., 2004; Nguyen et al., 2006; Sato et al., 2004). But, the precise mechanism in which its downstream effectors, Tcf/Lef factors, mediate these decisions is not known. Interestingly, the depletion of Tcf3 limits the ability of ESCs to differentiate (Pereira et al., 2006; Tam et al., 2008). Part of this is attributed to the relief of repression on Oct4 and Nanog, whereby their levels become induced. Comprehensive analyses of Tcf3 binding sites across the genome surprisingly revealed that this repressive factor share many common targets with Oct4, Sox2 and Nanog (Cole et al., 2008; Tam et al., 2008; see Figure 4A). The close proximity of these factors on co-occupied genes further suggests an integrative regulatory function, and many of their common targets have roles in ESC self-renewal. It appears conceivable that Tcf3 counters the effects of positive regulators on these genes as Tcf3 depletion induces the up-regulation of these targets (Tam et al., 2008). The conglomeration of data indicates that fine-tuning the dosage of ESC factors is a crucial component in transcription regulation (see Figure 4B). Currently, the interaction between activators and repressors on the same gene promoter is not well-understood, much less their complex dynamics on a global scale. The delicate balance between positive and negative inputs necessary to dictate the transcriptional output needs to be better established before quantitative evaluation and predictions of the transcription circuitry are possible (see Table 1). The recruitment of co-repressors, such as Grouchos/TLEs or activators such as β-catenin in the case of Tcf/Lef factors (Chen and Courey, 2000; Eastman and Grosschedl, 1999; Roose and Clevers, 1999), further confounds our understanding on how TFs act as molecular switches in a combinatorial manner. In-depth studies of interactions among transcription regulators and the global mapping of an increasing number of ESC factors are pertinent in the systems approach to dissecting the stem cell identity, as exemplified in a computational study model (Zhou et al., 2007).
3.4. Transcription factor pleiotrophy
Surprisingly, the role of Tcf3 extends beyond its control of pluripotency-associated genes. It also binds the promoters of homeodomain genes which include members of the Fox, Tbx and Sox gene families known to regulate early developmental programs (Buckingham et al., 2003; Kiefer, 2007; Plageman and Yutzey, 2005; Tam et al., 2008; Wijchers et al., 2006). Tcf3 appears necessary to prevent the premature activation of differentiation programs. Whereas polycomb group (PcG) proteins act as general transcription repressors that blocks the expression of a large cohort of developmental regulators (Boyer et al., 2006), Tcf3 binds to two categorically discrete sets of regulators in the same stage-specific context, i.e. undifferentiated ESCs. Although both sets of targets become de-repressed upon the loss of Tcf3, it remains unclear why Tcf3-deficient cells tend to self-renew rather than differentiate. A plausible explanation may be attributed to the functional dominance of ESC stemness-associated over differentiation-associated genes, as observed during somatic cell reprogramming by fusion with ESCs (Cowan et al., 2005; Tada et al., 2001). This pleiotropic control by a TF in a genome-wide manner is puzzling, and implies critical roles for repressors. Interestingly, it has also been demonstrated that Wnt signal activation can induce ESC-associated targets of Tcf3, possibly due to the replacement of co-repressors with the co-activator β-catenin which associates with Tcf3 (Cole et al., 2008). It, however, remains unresolved whether there is indeed a direct protein-protein interaction between Tcf3 and β-catenin upon Wnt stimulation, or that β-catenin may interact with other Tcf/Lef factors to activate common genes.
3.5. Functional validation of transcriptional targets
A major limitation of genome-wide studies is the generation of large interactome datasets that are not functionally tested in a biologically relevant manner. Technically, it is demanding to prove a causative relationship between gene occupancy and transcription activity one gene at a time. Although an alternative method is to correlate global transcript changes with factor activity for implying a direct transcription control, many studies surprisingly revealed that a large proportion of these gene targets do not appear directly regulated. Transcription factor over-expression or depletion does not necessarily alter the transcript levels of target genes. This, therefore, raises an important question of whether TF occupancy alone is sufficient to infer function in an overall network, or is the association with co-factors and protein complexes really the critical determinant. Furthermore, the epigenetic state of chromatin in altering permissiveness to transcription activity is another major consideration not discussed here (Azuara et al., 2006; Bernstein and Kellis, 2005; Bernstein et al., 2006; Guenther et al., 2007; Mikkelsen et al., 2007). One approach which clarifies the role of transcription targets is through gain- or loss-of-function studies to test if putative candidates are physiologically relevant. The availability of extensive gene expression libraries has made this type of validation possible (Chang et al., 2006; Paddison et al., 2004; Silva et al., 2005; Silva et al., 2004). It is now feasible to establish high-throughput platforms for introducing a library of over-expression or RNAi plasmid constructs into stem cells, accompanied by a suitable assay method for assessing gene function. For genetic studies in ESCs and gene function during animal development, the accessibility of gene-trap mouse ESC lines has made it easier to analyze gene functions in the animal (Hansen et al., 2003; Nord et al., 2006; Stryke et al., 2003; To et al., 2004). Hence, genomic discoveries create a framework which supports hypothesis-driven candidate gene function analysis.
4. The transcriptional landscape in somatic stem cells
4.1. Distinct transcription factors direct a common program
The global transcription landscape in many somatic stem cells is poorly understood. Part of this limitation is the inherent difficulty of obtaining a large number of homogenous bona fide somatic stem cells in vitro for analyses. Most somatic stem cells, unlike ESCs, arrest at the interphase of cell cycle and do not proliferate rapidly unless induced. Limited knowledge of the culture condition has prevented efficient expansion. Notwithstanding, unipotent and multipotent stem or progenitor cells of several tissues can be isolated and cultured. During myogenic differentiation, the convergence of lineage-defining TFs on a common set of targets amplifies the recurrent theme of a core transcription network. Precursor myoblast cells proceed through irreversible cell cycle arrest, followed by an increase in muscle genes expression. This process is orchestrated through a series of transcription networks governed by distinct myogenic TFs. Several basic helix-loop-helix (bHLH) family members play discrete roles. MyoD and Myf5 are necessary to specify the initial skeletal muscle lineage (Rudnicki et al., 1993), where the ectopic expression of MyoD alone is sufficient to force non-muscle cells to complete the myogenic program (Tapscott et al., 1988). The subsequent expression of MyoG causes the terminal differentiation of specified muscle cells (Hasty et al., 1993; Nabeshima et al., 1993), and Mrf4 appears to act as a specification as well as differentiation factor (Kassar-Duchossoy et al., 2004). In the skeletal muscle program, genome-wide identification of MyoD and MyoG binding sites revealed that both factors have distinct and sequential regulatory roles on a similar set of genes promoters (Cao et al., 2006; see Figure 2B). MyoD initiates chromatin remodeling but is insufficient for gene expression, and MyoG can only bind genes previously initiated by MyoD. The shared consensus DNA binding sequences of MyoD and MyoG further confirms their convergence (Berkes et al., 2004). This sustained temporal activation of similar targets by two stage-specific factors is surprising because previous evidence suggest that MyoD and MyoG act on separate sets of myogenic gene promoters thereby promoting a step-wise maturation (Asakura et al., 1993; Yutzey et al., 1990). The convergence of both factors on common targets could not have been revealed in limited candidate gene analyses, as a comprehensive approach to mapping the transcription landscape during myogenesis is necessary to resolve these differences.
4.2. Spatial and temporal expression of transcription factors
The specification of the vertebrate central nervous system is defined by neural progenitor/stem cells (NPCs/NSCs) which proliferate, self-renew, and give rise to neurogenic and gliogenic lineages. A few pro-neural genes which encode the bHLH TFs are necessary and sufficient to initiate the formation of neural lineages, and also to promote the generation of progenitors that are committed to differentiation (Guillemot, 1999). Neurogenins (Ngn1/2/3) and NeuroD are key regulators of vertebrate neurogenesis. Ngn1 or Ngn2 mutant mice lack cranial and spinal sensory ganglia and ventral spinal cord neurons (Fode et al., 1998; Ma et al., 1998; Ma et al., 1999), while NeuroD mutants manifest the loss of cerebellar and hippocampal granule cells, inner ear sensory neurons, and retinal photoreceptor cells (Kim et al., 2001; Miyata et al., 1999; Pennesi et al., 2003). The expression of pro-neural genes in NPCs is transient as these must be down-regulated before progenitors exit the proliferative zone of the neural tube and differentiate (Fode et al., 2000). Hence, pro-neural genes appear to promote complete neuronal differentiation through the induction of downstream regulators. The direct transcriptional targets of pro-neural genes has not been systematically defined, but indirect evidence in Xenopus suggests that Ngn1/2 and NeuroD may regulate many genes such as Nhlh1, NeuroD4 and Hes, which are related to neurogenesis (Seo et al., 2007). In pancreatic islet cells, NeuroD also appears to be a downstream mediator of Ngn activities (Huang et al., 2000).
Unlike ESC regulators which must remain constitutively elevated, the control of NPC fate by pro-neural genes occurs in two phases (Bertrand et al., 2002). The first involves a reversible selection of progenitors which are not committed to differentiation when pro-neural genes are expressed at a low basal level. This is followed by a second phase whereby elevated levels of pro-neural genes force an irreversible commitment of progenitors to differentiate. Although signaling pathways that include Notch, sonic hedgehog (Shh) and bone morphogenic protein (BMP) provide extrinsic cues for the initiation of pro-neural genes, it is likely that other positive feedback mechanisms mediated by downstream transcription targets help increase or maintain their appropriate expression in the selected progenitors. Subsequently, upon the induction of differentiation, lineage-restricted genes become elevated and direct the progenitors to adopt their committed fate. Even though pro-neural and neuronal-differentiation genes perform distinct functions, the structural similarities between them have raised the question of whether their disparate functions may be due to the consequences of stage-specific and spatial-dependent expression in the neural lineages, or intrinsic biochemical differences such as protein activity and/or stability (Bertrand et al., 2002). In the spinal cord, the spatial expression of neural bHLH proteins Math1, Ngn1 and Mash1 in distinct dorso-ventral progenitor domains is responsible for controlling the specification of interneuron subtypes (Gowan et al., 2001). Observations from myogenic differentiation appear to support the view that temporal expression of structurally related TFs plays a major role in defining the transcription program of either self-renewal or differentiation. Therefore, the expression specificity in a spatial and temporal context, rather than unique biochemical properties, of bHLH pro-neural genes may determine the control of differential target genes.
Several members of the Sox family of TFs also mark neural progenitor cells throughout the central nervous system, as well as maintaining self-renewing NPCs in vitro. The constitutive expression of Sox2 inhibits neuronal differentiation and results in the maintenance of progenitor characteristics, whereas its loss results in NPCs exiting cell cycle and the onset of neuronal differentiation (Graham et al., 2003). The multipotent NSCs obtained from mouse embryonic neuroepithelial cells express Sox2, along with the neural cell marker Nestin. Strikingly, Sox2 is also a determinant of ESC function. This underlying importance of the same factor in two stem cell types could suggest that either the temporal and lineage-specific expression of a TF determines their cell fate, or Sox2 could be a ‘stemness’ gene common to certain stem cells. Nevertheless, the lack of information on Sox2 transcription targets in NSCs hampers a direct comparison of the mechanisms which it may utilize in different stem cells.
4.3. A transcriptional link between hematopoiesis and leukemia?
During hematopoiesis, the establishment and maintenance of the entire blood system depends on rare hematopoietic stem cells (HSCs) residing in the bone marrow. The balance between self-renewal, and the differentiation of HSCs into lineage-restricted progenitors, and ultimately into diverse blood cell types, is intricately controlled through extracellular and intracellular signaling cues. These feed into the activation of regulators, which establish complex transcription networks. Briefly, HSCs can give rise to two major lineage-restricted progenitor cell types: the common myeloid progenitor which diversifies into myeloid cells comprised of monocytes, neutrophils, eosinophils, erythroids, mast cells and megakaryocytes; and the common lymphoid progenitor which produces the B and T lymphocytes (reviewed in (Orkin, 2000)). Several TFs maintain HSC self-renewal, whereas others specify differentiated hematopoietic lineages. The bHLH factor SCL, the SET-domain containing histone methyltransferase MLL and runt-domain Runx1 factors, are essential for production, survival and self-renewal of HSCs (Orkin, 2000). MLL and Runx1 are reported to regulate HoxB4 and Pu.1, respectively, whereby the loss of either factor blocks self-renewal (Diehl et al., 2007; Huang et al., 2008). Presumably, other yet unidentified downstream regulators may also be important. One lesson drawn from the identification HSC regulators is that many of these are concurrently mis-expressed in various human leukemias (Orkin and Zon, 2008). For instance, SCL is associated with T-cell acute leukemia, and MLL and Runx1 is associated with myeloid and lymphoid leukemias. The transcriptional control of genetic targets by these regulators during hematopoiesis and oncogenesis has not been forthcoming. It remains uncertain if a common factor does indeed controls similar self-renewal genes which is regulated in one cell type but deregulated in the other.
4.4. Co-factors and post-translation modifications impart transcriptional specificity
Important principles underlying the transcriptional control of erythropoiesis have emerged from mechanistic studies on the synergism and antagonism of factors involved in lineage transition. Gata1, the founding member of the GATA TF family is a determinant for common myeloid progenitor specification. Analyses of chromatin occupancy revealed that Gata1 occupies only a small proportion of high-affinity GATA motifs in the genome (Grass et al., 2006; Johnson et al., 2007a). Interestingly, Gata2, which contains a nearly identical dual zinc finger DNA binding domain, function uniquely to control distinct aspects of hematopoiesis (Lurie et al., 2008). Gata2 is expressed in HSCs to promote their proliferation, survival and function. Intriguingly, during erythropoiesis, Gata1 and Gata2 show reciprocal expression, with the levels of Gata1 increasing and Gata2 declining. Since both Gata1 and Gata2 could occupy similar target genes containing the GATA motif, how does the subsequent expression of Gata1 determine the activation of genes necessary for erythropoiesis? This selectivity appears to be partially imparted by the cell-specific co-regulator Friend of Gata1 (FOG-1) which mediates certain functions of Gata1 (Letting et al., 2004). Co-regulator proteins can be recruited to chromatin by sequence-specific TFs and catalyze chromatin modifications that control DNA accessibility and binding of the transcription machinery (Pal et al., 2004). However, the logic which dictates specific recognition of cognate Gata1 versus Gata2 targets, as well as their unique transcription output remains unclear. The question of how related factors are recruited to common versus distinct chromatin target sites needs to be resolved.
While the intrinsic differences in biochemical properties between related TFs are often under-appreciated, it could explain the phasic expression of related members which may perform unique and/or redundant functions. For example, the considerable protein stability of Gata1 compared with Gata2, conferred by the C-termini, provides an alternative explanation for selective target gene occupancy (Lurie et al., 2008). Gata1 is subjected to phosphorylation and acetylation, but it is unclear how these modifications affect Gata1 activity or function. The relevance of post-translation modifications to stem cell factors in defining genome-wide localization has not been examined. Although phosphorylation of the downstream effectors, Stat3 and Smad, of the Jak and BMP signaling pathways are essential for nuclear localization and DNA binding properties in ESCs, it is unclear how such modifications alter the binding dynamics of other stem cell factors on a global scale. There is emerging evidence that ESC factors such as Oct4 is subjected to sumoylation that enhances its stability and transactivation function (Wei et al., 2007). The Src family of tyrosine kinases has also been reported to be important for ESC self-renewal (Anneren et al., 2004), emphasizing the under-appreciated aspect of post-translation modifications in controlling transactivation activity and occupancy profile. Thus, differential stability of TFs can determine chromatin occupancy and help establish the appropriate genetic networks.
4.5. Surrogates to somatic stem cells?
Stem cells and cancer cells share several traits. Distinct from normal somatic cells which contribute to tissue function and homeostasis, both cell types are capable of sustained self-renewal in vitro and in vivo. The scarcity of cultured stem cells has, in part, led to the use of cancer cell lines such as Burkitt's lymphoma cells (Li et al., 2003), colorectal cancer cells (Hatzis et al., 2008), gliomas (Bruce et al., 2004), and T-cell acute lymphoblastic leukemic cells (Palomero et al., 2006) as surrogates for genome-wide TF location analyses. For example, the immortalized human non-neuronal T-lymphocyte cell line Jurkat is used to map binding sites for REST (Johnson et al., 2007b). Consistent with its current knowledge as a repressor of neuronal gene expression, a significant proportion of its targets is related to neurons and their development, synaptic transmission, and nervous system development (Johnson et al., 2007b). The oncogene c-Myc initially identified to be elevated in the lymph cells of patients suffering from Burkitt's lymphoma due to chromosomal translocations (Blum et al., 2004; Boxer and Dang, 2001), has been later discovered as one of the key factors for inducing pluripotent stem cells from somatic cells (Takahashi and Yamanaka, 2006). The pervasive importance of c-Myc in underlying stem cell and cancer cell functions has led to its genome-wide mapping in Burkitt's lymphoma cells and ESCs (Kim et al., 2008; Li et al., 2003). In both self-renewing populations, c-Myc is recruited to similar categories of genes that are involved in metabolism, cell growth and proliferation, and apoptosis. The resemblance, in which a TF may be expressed in different cell types but operate in an apparently similar context, thus provides compelling justifications for using cell lines as surrogates. However, it is imperative to note that surrogates provide at best a rough approximation of TF function. Even tissue stem cell lines arguably provide an estimation of their in vivo function owing to the effects of long-term culture adaptation and exposure to signals different from the niche context. On-going efforts in the generation of tandem affinity-tagged stem cell-specific TFs amendable to high quality immunoaffinity capture in transgenic animals constitutes the next step forward in unraveling transcriptional targets in the physiological context (Veraksa et al., 2005).
5. Reactivating pluripotency
Somatic cells are functionally specialized cells programmed to be in a stable state. In a landmark experiment, the somatic nucleus from Xenopus can be reverted to totipotency by an enucleated oocyte, and becomes capable of recapitulating complete animal development (Gurdon et al., 1958). Subsequently, the same phenomenon was recapitulated with the mammalian sheep somatic nucleus (Wilmut et al., 1997). The reversal of a differentiated cell to a developmentally more primitive state is termed ‘reprogramming’. Using an in vitro system, the generation of iPS cells from somatic cells through careful manipulation of specific molecules has been demonstrated (Takahashi and Yamanaka, 2006). By over-expressing a limited but variable combination of ESC-associated molecules comprised of Oct4, Sox2, c-Myc, n-Myc, Lin28, Nanog, Klf2 and Klf4, stem cells can be induced from embryonic and adult somatic cells such as fibroblasts, keratinocytes, liver and stomach epithelial cells of mouse and human (Aoi et al., 2008; Maherali et al., 2007; Meissner et al., 2007; Nakagawa et al., 2008; Park et al., 2008; Takahashi et al., 2007; Wernig et al., 2008a; Wernig et al., 2007; Wernig et al., 2008b; Yu et al., 2007b). These karyotypically normal cells exhibit morphology, growth properties and molecular signatures similar to ESCs. Notably, iPS cells can contribute to all germ layers of the chimeric mouse, and are capable of germ-line transmission. Here, an understanding of the transcription targets of these ‘reprogramming factors’ has provided useful insights into the mechanism for pluripotency reactivation.
In ESCs, Oct4 and Sox2 bind and regulate downstream genes, which corroboratively contribute to reverse the differentiated cell phenotype, through controlling a host of genes responsible for maintaining pluripotency or inhibiting differentiation described previously. The relevance of c-Myc and Klf4 targets appear to be more speculative. From recent studies, the role of c-Myc in sustaining self-renewal may be attributed to its many targets that enhance proliferation, negatively regulate differentiation, cellular transformation, and regulation of chromosome accessibility to other factors (Adhikary and Eilers, 2005; Niwa, 2007; Takahashi and Yamanaka, 2006). The permissiveness of chromatin to TF binding and transcription activity is partly determined by histone modifications. The specific covalent modifications on the histone moieties of nucleosomes can be correlated to the transcription activity of genes in either promoting or repressing their expression (reviewed in (Strahl and Allis, 2000)). In ESCs, a large set of developmentally important genes that are silent but activated upon differentiation carries the bivalent marks, consisting of the activating histone 3 lysine 4 trimethylation (H3K4me3) and the repressive H3K27me3 modifications (Bernstein et al., 2006; Mikkelsen et al., 2007). This suggests lineage-specifying genes are primed for activation upon ESC differentiation.
A global assessment of the bivalent histone marks of c-Myc target promoters has revealed that the majority (95%) bear the H3K4me3 mark (Kim et al., 2008), suggestive of a relationship between c-Myc occupancy and histone modifications (Guccione et al., 2006). Indeed, these target genes are more frequently expressed in ESCs, compared to other factors. The data indicates that c-Myc occupancy is associated with global epigenetic modification of the chromatin at its targets, distinct from the role of other core factors which may manifest bivalent domains comprising of both H3K4me3 and H3K27me3 marks (Kim et al., 2008). This latter data is coherent with observations in human and mouse ESCs where there is considerable overlap between H3K4me3 and H3K27me3 loci across the pluripotent genomes (Azuara et al., 2006; Bernstein et al., 2006; Pan et al., 2007; Zhao et al., 2007). In addition to histone methylation, c-Myc may also potentially modify the chromatin architecture to be more permissive for the binding of Oct4 and Sox2, through its regulation of histone acetyltransferase complexes that induce global histone acetylation (Takahashi and Yamanaka, 2006). The role of Klf4 in ESCs and in reprogramming is less clear, but circumstantial observations hints that it could act as a tumor-suppressor gene in iPS cells. Klf4 inhibits cell growth in mouse (Shields et al., 1996), and can reduce the tumorigenicity of colon cancer cells in humans (Dang et al., 2003). This may be correlated to the observations that during the reprogramming process, bona fide iPS cells are sparsely scattered amidst large numbers of transformed colonies. Presumably, Klf4 could act as a selector for truly reprogrammed cells over transformed or incompletely reprogrammed cells which are deregulated in proliferation control. Klf4 may also act via repression of p53 which leads to the activation Nanog and other ESC-associated genes (Rowland et al., 2005; Takahashi and Yamanaka, 2006). The occupancy of Klf4 on Oct4, Sox2 and other common downstream targets that include Nanog and c-Myc has revealed that it is an upstream regulator of positive feed-forward loops that sustain high levels of the activated factors. Interestingly, Klf2 can replace Klf4 in reprogramming (Nakagawa et al., 2008), and it has been shown that Klf2, Klf4 and Klf5 bind to similar targets in ESCs (Jiang et al., 2008). The amenability in which the combinatorial use of reprogramming factors results in the generation of iPS cells has raised interesting speculations pertaining to the plasticity of reprogramming. On the one hand, certain TFs appear mutually replaceable to target common genes possibly through motif recognition; on the other hand, the activation of distinct but functionally similar target genes may, in concert, results in induced pluripotency.
Further attempts to dissect the mechanism of reprogramming through examination of partially reprogrammed cells have provided strong evidence of a direct connection between transcription regulation and epigenetic modification during the transition of cellular states. Whereas fully reprogrammed cells manifest gene expression and epigenetic profiles that are similar to ESCs, partially reprogrammed cell lines do not completely repress lineage-specifying TFs and show DNA hypermethylation at the loci of pluripotency-related genes (Mikkelsen et al., 2008). Strikingly, silencing of specific lineage-specifying TFs and treatment with DNA methyltransferase inhibitors help improve the efficiency of the reprogramming process (Mikkelsen et al., 2008), supporting genome-wide observations that methylation of CpGs are dynamic epigenetic marks that are subject to extensive changes during differentiation (Meissner et al., 2008), and need to be appropriately reversed at specific genes during reprogramming. Presumably, partially reprogrammed entities are trapped in the intermediate state as a result of either the inappropriate silencing of pluripotency factors or the mis-expression of differentiation-associated genes. A better understanding of the role of lineage-specifying TFs, and the requirements for epigenetic landscape that include DNA and histone modifications at specific gene loci, may facilitate in reprogramming differentiated cells to somatic stem cells which are clinically relevant.
6. Cell type-dependent regulation of distinct circuitries
The molecular mechanism for the specification of embryonic germ layers and extra-embryonic lineages during pre-implantation development is an important area that is not well-understood. With genome-wide TF localization analyses, precise and detailed maps can provide a comprehensive view of the molecular circuitry governing embryonic development. Although much effort has focused on how different TFs act singly or collectively to regulate extensive networks (Boyer et al., 2005; Kim et al., 2008; Loh et al., 2006), there is scant knowledge on how the same TF may behave in different stem or progenitor cells. This is largely due to the paucity of identified molecules that are common in more than one stem cell types, but yet retain discrete functional significance in each lineage (Fortunel et al., 2003).
In several cell types, the mapping of lineage-determining TFs has placed them at the top of transcription hierarchies. These factors are largely auto-regulatory, but also orchestrate the expression of downstream effectors. These effectors may independently or collaborate with other factors to control the cellular response. The observation that a TF controls its own expression is not surprising as auto-regulatory loop appears to be a general feature of ‘master’ regulators (Odom et al., 2006). For example, HNF TFs are required for the normal function of liver and pancreatic islets. In human hepatocytes, HNF1α binds a substantial number of genes related to hepatocyte biochemistry such as gluconeogenesis, carbohydrate synthesis, lipid metabolism and detoxification. This network also includes HNF4α and HNF6 which co-occupy the promoters of one another, thus forming a core network (Odom et al., 2004).
6.1. Lineage-specific recruitment
One central theme has been the prevalent observation that a lineage-specific factor controls of a discrete transcription network. Recently, we have identified Sall4 as a key regulator common to two embryo-derived stem cell lines that are developmentally connected (accepted, Cell Stem Cell). Our deciphering of Sall4 targets is timely as integrative studies from the mapping of multiple TFs has identified Sall4 to be a major regulatory node in ESCs (Kim et al., 2008). Many Sall4-regulated genes are known to play important roles in maintaining ESC pluripotency. Of significance, many homeodomain protein promoters occupied by multiple factors such as Nanog, Sox2, Oct4 and Klf4 (Kim et al., 2008), are also regulated by Sall4. These findings thus integrate Sall4 as a major component of the core ESC circuitry.
In addition to defining its role in ESCs, we demonstrated the importance of Sall4 function in the primitive endoderm stem cell derivative, XEN cells. Our studies reveal Sall4 as an upstream activator of key lineage-defining genes in the primitive endoderm. Gata6 and Gata4 are two of the earliest determinants of the primitive endoderm. However, other factors appear necessary for the initial primitive endoderm specification as Gata6 and Gata4 mutants do not exhibit extra-embryonic endoderm defects until several days after blastocyst formation (Ralston and Rossant, 2005). The discovery that Sall4 lies upstream of Gata6 and Gata4, and drives the expression of other lineage-determining genes, provides a plausible reasoning for why the loss of either factor does not lead to a complete disruption of primitive endoderm formation, but only affects visceral endoderm differentiation at later stages (Koutsourakis et al., 1999; Morrisey et al., 1998; Ralston and Rossant, 2005; Soudais et al., 1995). These observations thus support Sall4's role as a central player in primitive endoderm and XEN cell maintenance.
Genome-wide mapping of the same TF in two distinct stem cell lines has revealed several novel findings (see Table 1). Firstly, Sall4 regulates distinct genes in the different cell types. In ESCs, pluripotency-associated genes predominate; whereas in XEN cells, endoderm lineage-associated factors that include Gata4, Gata6, Sox7 and Sox17 are targets (see Figure 2C). Secondly, while the recruitment of Sall4 and its interacting partners appear necessary to coordinate the expression of a subset of genes in establishing a specific lineage program in one cell type, it is equally important that the promoters of the same subset of genes become inaccessible to Sall4 in another stem cell type to ensure the complete absence of Sall4 regulatory input. This study illustrates and provides a basis for future investigations into how other similar factors could have unique roles in more than one progenitor cell type, as well as further our understanding on the genetic and epigenetic bases for self-renewal and lineage-potency in stem cells.
6.2. Cofactor-mediated differential recruitment
The interaction of a sequence-specific TF with co-factors plays a contributory role in its differential recruitment. Sall4 physically interacts with several factors that include Nanog, Dax1, Nac1, Zfp281 and Oct4 as protein complexes in ESCs (Wang et al., 2006; Wu et al., 2006). However, the identity of its partners in XEN cells is unknown. Whether the recruitment to specific promoters in different cell types is partially mediated by other sequence-specific co-factors remains to be tested. Notably, the reliance on co-factors for conferring recruitment specificity appears to be a recurrent theme that has been observed in non-stem cells. For example, FoxA1 (HNF3α), a member of the Forkhead family of winged-helix TFs, is involved in the organogenesis of liver, kidney, prostate, and mammary gland (Friedman and Kaestner, 2006). Interestingly, the high expression of FoxA1 in tumors arising from the prostate and breast has led to speculation on the similarities or differences of its transcription dynamics between these cell types (Lin et al., 2002; Mirosevich et al., 2006). In breast cancer cells, FoxA1 behaves as a pioneer factor that mediates estrogen receptor α (ERα) recruitment to cis-regulatory elements (Carroll et al., 2005; Laganiere et al., 2005), whereas it interacts with the androgen receptor (AR) in prostate cancer cells (Gao et al., 2003). A comparison of the FoxA1 binding loci between the breast and prostate cancer cells has shown that FoxA1 is recruited to largely different sites across the genome (Lupien et al., 2008). Coherent with similar observations made in ESC and XEN cells, FoxA1 appears to regulate discrete transcription programs as a result of lineage-specific recruitment (see Table 1). While the transcription landscape of ESC factors is primarily associated with promoter occupancy, the ERα or AR-dependent recruitment of FoxA1 occurs at enhancers (Lupien et al., 2008). The Forkhead motif is enriched at the binding loci both cell types, but the recognition motifs for ERα and AR are specifically enriched in FoxA1 binding loci unique to either breast or prostate cancer cells, respectively. The extensive occupancy of TFs at enhancers on a global scale supports the notion of enhanceosomes which may be prevalent features in many cell types.
The transcription switch in the regulation of differential gene sets following cellular perturbation provides an interesting framework for testing the dynamicity of factor localization to the chromatin (see Table 1), as well as the requirements of co-factors and/or epigenetic permissiveness. For example, NF-κB, a key mediator of inflammation, responds to lipopolysaccharide (LPS) stimulation in monocytic cells. On a global scale, NF-κB recruits E2F1 to fully activate the transcription of NF-κB target genes upon LPS induction, whereby this effect could be abolished through the depletion of E2F1 by RNAi (Lim et al., 2007). This fluid nature of TF recruitment raises interesting questions of whether forced expression of co-factors not naturally present in the target cell could induce the binding to, and expression of alternate genes, and elicit a change in phenotype. Through this manner, the forced ectopic expression of ESC TFs in somatic cells has been shown to dramatically alter the transcription landscape and cellular identity, leading to the induction of pluripotent cells.
7. The integration of microRNAs in transcription circuits
The governance of stem cell identity is not limited to transcription networks. Post-transcriptional regulatory mechanisms modulate the expression of many genes, emphasized by the many points of control in eukaryotes (Hollams et al., 2002). It is now apparent that a class of non-coding RNAs, microRNAs, modulates mRNA decay and translation rates (Seitz et al., 2004). MicroRNAs are necessary for many aspects of animal development and stem cell maintenance. For instance, the miR-17∼92 cluster which is highly abundant in mouse ESCs has extensive roles in the development of the heart, lungs, and immune system, as well as tumorigenesis (Koralov et al., 2008; Ventura et al., 2008; Xiao et al., 2008; Yu et al., 2007a). The skin microRNA, miR-203, represses p63 and promotes differentiation of stratified epithelial stem cells (Yi et al., 2008b). In mouse ESCs, miR-134 and miR-1 can induce differentiation into the neuroectoderm and cardiac muscle lineage, respectively (Ivey et al., 2008; Li and Gregory, 2008; Tay et al., 2008). The importance of microRNAs in stem cells is further highlighted by the observation that ESCs lacking the microRNA processing machinery comprised of either Dicer or Drosha cannot differentiate into most lineages (Kanellopoulou et al., 2005; Murchison et al., 2005; Wang et al., 2007).
Despite our increasing knowledge of microRNA functions and their regulation of target gene translation, there is a dearth of information regarding the control of their expression. Unlike most protein-coding transcription units which contain obvious transcription start sites and other initiation landmarks, microRNA promoters are not well-characterized. The occupancy of a TF on a microRNA gene is at best determined by its proximity to infer some regulatory function. In human ESCs, at least 14 microRNA loci are bound closely by Oct4, Sox2 or Nanog, where miR-137 and miR-301, are co-occupied by all three TFs (Boyer et al., 2005). In the mouse counterpart, five microRNA loci are bound by Oct4 or Nanog, where miR-296 and miR-302 are co-occupied by both (Loh et al., 2006). These two microRNAs are highly expressed in ESCs (Houbaviy et al., 2003), and thought to contribute towards ESC function. Interestingly, the mouse miR-302a-e cluster is the homologue of the human miR-372/3 family which is highly abundant in human ESCs, and acts as oncogenes in testicular germ cell tumors. When introduced into primary human cells, miR-372/3 can induce cellular transformation (Voorhoeve et al., 2006), suggesting a common role for this microRNA family in cell proliferation. Although many of these ESC-specific microRNAs appear to be under the control of key TFs, there is surprisingly little demonstration of their direct regulation.
The repressor REST is observed to bind several microRNA promoters which contain REST regulatory sequences in ESCs (Singh et al., 2008). One of these microRNAs, miR-21, has been previously characterized as an ‘oncomir’ that promotes transformation in cells by targeting tumor suppressors (Asangani et al., 2008; Lu et al., 2008; Zhu et al., 2007; Zhu et al., 2008). Its levels also appear elevated in certain primary cancer cells (Meng et al., 2007; Slaby et al., 2007). While REST regulates ESC pluripotency possibly through its silencing of developmental genes associated with the neural lineage (Ballas et al., 2005; Bruce et al., 2004; Kim et al., 2008), it also blocks the expression of target microRNAs which may promote differentiation (Singh et al., 2008). Elevating the level of miR-21 reduced the self-renewal capacity of ESCs (Singh et al., 2008); and it is proposed that miR-21 may potentially alter the proteins levels of its computationally predicted targets, Sox2 and Nanog, although this has yet been demonstrated. Parallel work on miR-134 has shown that it suppresses the translation of ESC factors Nanog and LRH1, thereby leading to cellular differentiation (Tay et al., 2008). It would indeed be fascinating if miR-21 feeds back into the transcription circuitry by suppressing the activity of its target TFs. The transcriptional control of microRNAs and their re-integration into the circuit presents a novel facet of gene regulatory network that is currently not well-elucidated. There is emerging evidence that microRNAs are integral components of transcription circuitries, both regulating, as well as being regulated by, TFs (see Figure 5). Computation models predict that microRNA-mediated feedback and feed-forward loops are prevalent (Tsang et al., 2007). The TF input can either positively or negatively co-regulate a microRNA and its targets simultaneously, thereby enhancing the robustness of gene regulation and stability. This may be particularly important for reinforcing the gene expression program of differentiated cellular states (Tsang et al., 2007; Yoo and Greenwald, 2005).
The recent genome-wide analyses of multiple TF occupancy could help reveal microRNAs bound by multiple factors (Chen et al., 2008; Kim et al., 2008). It is interesting to test whether the distinctive classes of factors comprising of Oct4/Sox2/Nanog and n-Myc/c-Myc/Rex1 could regulate delineating classes of microRNAs. If this is true, it would then suggest that different classes of microRNAs perform specific roles that complement their upstream regulators. From a clinical perspective, the set of microRNAs bound by reprogramming factors is potentially important to examine. It remains to be tested whether any of these microRNA targets can replace or enhance TF-mediated reprogramming of somatic cells into iPS cells. The chemical simplicity and ease of synthesis of microRNAs are key advantages in a small molecule approach to generate medically useful stem cells.
In epithelial stem cells where p63 maintains the undifferentiated state (McKeon, 2004; Senoo et al., 2007), the interaction of miR-203 and p63 in repressing ‘stemness’ and inducing differentiation into stratified epithelium has been shown (Yi et al., 2008b). However, the manner in which p63 could trigger other microRNAs which help maintain undifferentiated stem cells is not known. Like transcription repressors, microRNAs are integral nodes of many gene regulatory networks. While microRNAs appear to predominantly operate through a repression mechanism, their functions in the context of networks need not be repressive (Tsang et al., 2007). Presumably, microRNAs could suppress certain TFs, leading to the activation of a second order hierarchy that initiates the next transcription program. The identification of microRNA signatures specific to many stem cell types such as HSCs, mammary and lung epithelial progenitor cells, and mesenchymal stem cells (Hatfield and Ruohola-Baker, 2008; Ibarra et al., 2007; Lakshmipathy and Hart, 2008; Liao et al., 2008; Lu et al., 2007), has provided the opportunity to intersect the regulatory networks of transcription regulators with microRNA and their target genes.
8. Perspectives
Investigations into transcription regulation have progressed from the elucidation of very limited gene interaction networks to the mapping of complex extensive interactions on a genome-wide scale. This is, in part, facilitated by the employment of high-throughput technologies in detecting and analyzing TF binding sites. Knowledge of how dominant factors control gene expression in stem cells and the progression of tissue lineage commitment during development is particularly important. As the stage-wise transition of pluripotency to multipotency and finally terminally differentiated cells requires the timely activation of a cascade of unique transcription programs, the identification of key regulators in the hierarchy and their targets provides valuable clues into the manipulation of stem cells. Two aspects of this manipulation predominates the field of stem cell biology. Firstly, uncovering the transcription program of a differentiated cell type can provide information on directing the stem cell program to proceed towards that of the desired cell type. Understanding how to mature ESCs into stable functional somatic cells is the key to generating useful cell types for transplantation. Secondly, the derivation of patient-specific transplantable cells is a major challenge. Obtaining patient-specific ESCs has proved surmountable and has raised ethical concern. Hence, unraveling the somatic stem cell program can aid the reprogramming of terminal differentiated cells into all the various somatic stem cell types. This can potentially bypass the need for extensive differentiation regimes that would be required for the effective use of ESCs.
The observation that certain key transcription regulators previously thought to define a particular lineage is also abundantly expressed in other cell lineages is not new. As intrinsic determinants of the cellular identity, TFs provide an entry point for uncovering how stem or progenitor cells attain their phenotype, and how lineage-specific differentiation is programmed. However, insights into the mechanism for the differential function of these TFs are critically lacking. The simple explanation that a single factor controls a small handful of divergent lineage-restricted genes necessary for establishing distinct cellular identities does not satisfactorily resolve a complex developmental problem. The genome-wide identification of TF binding targets has brought us one step closer in our attempt to deconstruct the regulatory mechanism of stem cells. With the elucidation of complex gene regulatory networks, the field is only starting to reveal its secrets. Much of the on-going challenges will lie in our ability to dissect the mechanistic interaction of TFs with the chromatin. In particular, how the epigenetic state determines the specificity of factor recruitment, the requirement and function of co-regulators, the dissection of a combinatorial code for transcription output, as well as the activity and biochemical properties of TFs, will be key areas to study. Arguably, pioneering work in understanding the comprehensive stem cell regulatory network will form the basis for probing the greater depths of developmental biology, and our ontogeny.
Acknowledgements
This work is supported by the Agency for Science, Technology and Research (Singapore) and the Singapore Stem Cell Consortium grant (SSCC-06-03). The work is also partially supported by National Institutes of Health (NIH) grants to B.L. (DK47636 and AI54973). We are grateful to Leah Vardy and Yuin-Han Loh in the Genome Institute of Singapore for critical comments.