
1. Introduction:
Dual SMAD inhibition takes a confluent, feeder free culture of hPSCs and rapidly differentiates them into early neurectoderm (Chambers et al., 2009). This rapid differentiation is caused by blocking the two signaling pathways that utilize SMADs for transduction: BMP and TGFB. Oct4 is extinguished and Pax6 expression has begun by around day 7–8, depending on the line used. This neurectoderm can be passaged to become rosettes or can be patterning to become many other types of neural cells (for example, see Fasano et al., 2010).
2. Flowchart:
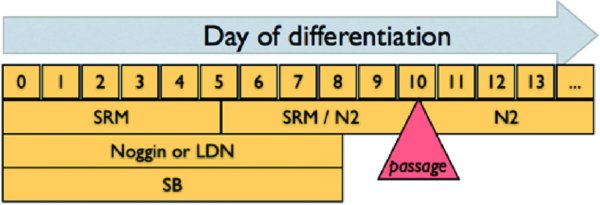
2.1. Creating a confluent hPSC monolayer for neural induction
-
Aspirate hPSC media and add minimal Accutase to coat the dish. [1 ml for a 6 well dish, 2 ml for a 6 cm dish, or 5 ml for a 10 cm dish. There is no need for a PBS wash.]
-
Place in the incubator (37∘C) for about 30 minutes - colonies will become single cells.
-
Pipette cells a few times to make a single cell suspension. Filter through a 45 μm strainer to remove any remaining clumps.
-
Wash and centrifuge cells (200×g, 5 min) two times in hPSC media to remove Accutase.
-
While washing, prepare Matrigel-coated dishes. Add 19 ml of hPSC media to a nearly-thawed Matrigel aliquot and pipette until completely thawed and homogeneous.
-
Work quickly and do not let the Matrigel warm up or it will polymerize.
-
After washing hPSCs, resuspend the cells in hPSC media with 10 μM Y-27632 and place on a gelatin-coated dish of the same size as used to grow the hPSCs in step 1.
-
Place dish in incubator (37∘C) for 30 min.
-
Collect the non-adherent cells and gently wash the dish with hPSC media with 10 μM Y-27632 and centrifuge the cells (200×g for 5 min).
-
hPSCs are rounded and therefore non-adherent. Contaminating MEFs are flat and more adherent. This step eliminates many MEFs by leaving them stuck to the dish.
-
Resuspend the cells in complete conditioned media (cCM) with 10 μM Y-27632.
-
Determine the cell concentration using a hemocytometer and add cCM with 10 μM Y-27632 to the appropriate cell concentration for desired density (see chart at end).
-
Aspirate the Matrigel solution and plate cells directly on dish. There is no need to wash the dish though some do.
-
Twenty-four hours after plating, aspirate media and add fresh cCM with 10 μM Y-27632.
-
Two days after plating, aspirate the media and add fresh cCM - from this point on, Y-27632 is no longer necessary.
-
Cells can be maintained for additional days in cCM until the desired density is reached (∼90-95% for CNS or about 60% for a mixture of neural crest and CNS).
2.2. Neural induction
-
To initiate differentiation, add SRM containing 10 μM SB431542 and 200 ng/ml Noggin.
-
Defined as Day 0 of the induction. 100 nM LDN 193189 can be used instead of Noggin for the protocol as well although we do not currently understand possible downstream changes in cell fate.
-
On day 1 of differentiation, aspirate the SRM and add fresh SRM with 10 μM SB431542 and 200 ng/ml Noggin.
-
On day 2 of differentiation, aspirate the SRM and add fresh SRM with 10 μM SB431542 and 200 ng/ml Noggin.
-
On day 4, aspirate the SRM and add a mixture of SRM/N2 media (3:1) with 10 μM SB431542 and 200 ng/ml Noggin (final concentration).
-
On day 6, aspirate the SRM and add a mixture of SRM/N2 (1:1) with 10 μM SB431542 and 200 ng/ml Noggin (final concentration).
-
On day 8, aspirate the SRM and add a mixture of SRM/N2 (1:3) with 10 μM SB431542 and 200 ng/ml Noggin (final concentration).
-
On day 10, cells can be passaged en bloc (variation 1) or as single cells (variation 2) onto Matrigel-coated dishes.
2.3. Mechanical dissociation (variation 1)
-
Mechanically dissociate thickened neurectoderm using a 200 μL pipette into small pieces, or cut monolayer of cells using a StemProEZ Passage tool (Invitrogen)
-
Plate the blocks of tissue onto Matrigel-coated dishes in N2 containing the appropriate growth factors at 2:1 or 3:1. Note that this is 2:1, not 1 : 2 - you want less surface area because not all of the pieces will attach and you need very high cell density to get good progression of CNS neural cells.
2.4. Single cell passage (variation 2)
-
Aspirate differentiation media and add minimal Accutase to coat the dish and let sit at 37∘C until all cells are rendered to single cells (approximately 30 minutes).
-
Avoiding bubbles, triturate the cells in the dish using a pipette with additional N2 media until the cells are in single cell suspension and filter using a 45 μm cell strainer.
-
Wash and centrifuge cells (200×g for 5 minutes) twice in N2 media.
-
Resuspend the cells in N2 media.
-
Determine the cell concentration using a hemocytometer.
-
Resuspend the cells in N2 media to a cell concentration of 5 × 106/mL.
-
Prepare Matrigel-coated dish by carefully aspirating all liquid from the dish, taking care not to touch the surface. Let dish dry for 15 minutes before plating drops.
-
Spot plate with 20 μL drops of the cell suspension (1×105 cells) and let stand in the hood for 20 minutes before slowly adding N2 containing the appropriate growth factors. Move dish carefully to the incubator.
2.5. Neuronal differentiation
-
Minimal supportive media for differentiating neural cells into neurons is N2 containing 20 ng/ mL of BDNF and 200 μM ascorbic acid. Differentiations are usually carried out for at least one (and sometimes two) weeks with media changes every two to three days. 2. To enrich for motor neurons, 20 ng/mL BDNF, 200 μM ascorbic acid, 50 ng/mL SHH (C25II) and 1 μM retinoic acid are added to the N2 base medium.
-
To enrich for dopamine neurons, 20 ng/mL BDNF, 200 μM ascorbic acid, 50 ng/mL SHH (C25II) and 100 ng/mL FGF8 are added to the base N2 media for one week, followed by 20 ng/ mL BDNF, 200 μM ascorbic acid, 20 ng/mL GDNF, 1 ng/mL TGF-B3, and 500 μM cAMP for all subsequent weeks.
-
To enrich for rosettes, use high density replating, 50 ng/ml SHH (C25II) and 100 ng/ml FGF8 and 20 ng/ml BDNF. Materials:
-
Matrigel Basement Membrane Matrix (BD Bioscience; cat. no. 354234: we only use lots that contain over 10 mg/ml protein). Thaw the frozen vial of Matrigel on ice overnight in a 4∘C refrigerator. Prepare 1 mL aliquots in a 50 mL centrifuge tube using chilled pipettes and freeze at −20∘C. Matrigel must be thawed slowly to prevent gelatinization. Chilled pipettes and 50 mL centrifuge tubes should be used when making aliquots of the Matrigel.
3. Materials:
3.1. MEF media
-
900 ml DMEM (Invitrogen cat.# 11965-118) 100 ml FBS (Invitrogen, cat.# 26140-095)
-
Filter sterilize.
3.2. hPSC media
-
790 ml DMEM:F12 (Invitrogen, cat.# 11330-032)
-
200 ml Knockout serum replacement (SRM; Invitrogen, cat. no. 10828-028)
-
5 ml L-glutamine (200 mM, Invitrogen, cat.# 25030-081)
-
10 mL MEM non-essential amino acids (MEM NEAA; Invitrogen, cat.# 11140-050)
-
1 mL of 2-mercaptoethanol (Invitrogen, cat.# 21985-023)
-
The medium is sterile filtered in the hood and FGF2 is added after filtration to a final concentration of 6 ng/ml.
3.3. SRM media
820 mL of Knockout DMEM (Invitrogen; cat. no. 10829-018), 150 mL Knockout Serum Replacement (Invitrogen, cat. no. 10828-028), 10 mL L-glutamine (200 mM, Invitrogen, cat. no. 25030-081), 10 mL MEM NEAA (Invitrogen, cat. no. 11140-050), and 1 mL of 2-mercaptoethanol (Invitrogen, cat. no. 21985-023).
3.4. MEF conditioned media (CM)
MEF CM is harvested from MEF coated flasks. MEFs are plated at a density of 50,000 cells/cm2 in a T225 flask in MEF media. The next day, the cells are washed once with PBS before adding 100 mL of hESC media. Incubate media with MEFs for 24 hours before removal. The medium is now known as “conditioned media” (CM) and can be directly used or stored at 4∘C for less than two weeks. Additional hESC media can be conditioned daily for up to ten days on the same flask of feeders. Just before using, FGF2 is added to CM to a final concentration of 10 ng/mL, hereafter called complete CM (cCM).
3.5. N2 media
N2 media contains DMEM/ F12 powder (Gibco/Invitrogen, cat no. 12500-062) in 550 mL of distilled water. 1.55 g of glucose (Sigma, cat. no. G7021), 2.00 g of sodium bicarbonate (Sigma, cat. no. S5761), putrescine (1 ml aliquot of 1.61 g dissolved in 100 mL of distilled water; Sigma, cat. no. P5780), progesterone (20 uL aliquot of 0.032g dissolved in 100 mL 100% ethanol; Sigma, cat. no. P8783), sodium selenite (60 uL aliquot of 0.5 mM solution in distilled water; Bioshop Canada, cat. no. SEL888), and 100 mg of transferrin (Celliance/Millipore, cat. no. 4452-01) are added. 25 mg of powdered insulin (Sigma, cat. no. I6634) is added to 10 mL of 5 mM NaOH and is shaken until completely dissolved. The solubilized insulin is added to the media, and double-distilled water (with a resistance of 18.2 MΩ) is added to a final volume of 1000 mL before sterile filtration.
***Recently, the Studer and Tomishima labs have had problems with the supply chain for this media. This has caused us to explore new sources, and many are now using Neurobasal with N2 supplements and B27 (without retinoic acid) and L-glutamine.
3.6. FAQs:
Q: I am having problems passaging at day 10 – what's wrong?
A: You have to keep a very high cell density for passage. Low densities do not survive well and will not rosette, particularly for the single cell passage. It might help to use Y-27632 to promote survival.
Q: I have seen many different cell densities for the start of neural induction. What's the right number?
A: Basically, you need to have confluent PSCs. We used to passage as singles and expand them in place before differentiation, but most of us now plate at saturating densities (200,000 cells/cm2), allow the cells to recover overnight, then induce differentiation. It might also be possible to induce differentiation at the replating point but I have not tried this yet.
Q: N2 is hard. Is there an easier way?
A: Maybe. Most people around here have switched to NeuroBasal with B27 (no RA) and N2 supplements (Stem Cell Technologies), L-glut and glucose as an alternative. This has seemingly fixed our inconsistencies with some differentiation protocols, but your mileage might vary. We do not yet have a clear understanding of what this could do to downstream differentiations.
References
Last revised March 28, 2012. Published June 10, 2012. This chapter should be cited as: Tomishima M., Neural induction - Dual SMAD inhibition (June 10, 2012), StemBook, ed. The Stem Cell Research Community, StemBook, doi/10.3824/stembook.1.57.1, https://www.stembook.org.
